
SHOCK STATES IN ACUTE CARE SURGERY
Shock is the clinical end result of inadequate tissue perfusion. The currently accepted concept was first described in 1918 by Walter B. Cannon.1 There are multiple causes of shock; however, the common pathway is an imbalance in oxygen delivery and utilization, and ultimately cellular dysfunction. Cellular hypoxia induces the production of inflammatory mediators that may further compromise tissue perfusion through changes in the microvasculature. If this vicious cycle is not interrupted, multiorgan failure (MOF) and death result.2 The clinical manifestations of shock are due to the autonomic neurohumoral responses to hypoperfusion and the resulting organ dysfunction.
This chapter describes the pathogenesis of shock and discusses the diagnostic and therapeutic options in current management.
PATHOGENESIS AND ORGAN RESPONSE TO SHOCK
At the cellular level, shock results in cellular hypoxia. Initially, the injury to cells is reversible; however, if it is not corrected, cell death ensues. The hallmarks of reversible injury are reduced oxidative phosphorylation with subsequent depletion of energy stores in the form of adenosine triphospate (ATP) and cellular swelling caused by failure of the sodium pump. In addition, massive influx of calcium occurs due to failure of the calcium pump, causing mitochondrial dysfunction. When injury becomes irreversible, the plasma membrane is disrupted; lysosomal enzymes enter the cytoplasm and digest the cell, resulting in necrosis.
Cellular hypoxia also predisposes to reperfusion injury, where new damaging processes are set in motion, causing the death of cells that might have recovered otherwise. Increased amounts of reactive oxygen and nitrogen species are generated from parenchymal and endothelial cells as well as infiltrating leucocytes.3 These free radicals may be produced as a result of mitochondrial damage and incomplete reduction of oxygen. Endogenous cellular antioxidant mechanisms are compromised, favoring the accumulation of free radicals. Increased calcium may also enter the reperfused cells, damaging mitochondria and producing further free radicals. Hypoxic cells produce cytokines and increased expression of adhesion molecules, which recruit circulating neutrophils to reperfused tissue. The resulting inflammation generates additional tissue injury. Activation of the complement system also contributes to reperfusion injury. Select IgM antibodies are predisposed to deposit in hypoxic tissue, and when blood flow is resumed, complement proteins bind to the deposited antibodies, thus causing activation and subsequent inflammation and cell injury.
Cardiovascular Response
Decreased tissue perfusion results in the release of epinephrine and norepinephrine from the adrenal medulla due to decreased afferent impulses from the arterial baroreceptors. Systemic vascular resistance (SVR) rises to maintain adequate perfusion of the heart and brain, at the expense of perfusion to the skin, kidneys, and, primarily, the gastrointestinal tract via splanchnic vasoconstriction. This peripheral vasoconstriction occurs primarily in the arterioles, mediated by α1 receptors. However, vasoconstriction also occurs in the precapillary and postcapillary sphincters and the small veins and venules. This results in a reduced hydrostatic pressure distal to the precapillary sphincter that leads to reabsorption of interstitial fluid into the vascular space. This reabsorption functions to restore intravascular volume and is known as transcapillary refill.
Cardiac output (CO), the product of stroke volume and heart rate (HR), is a major determinant of tissue perfusion. Decreased intravascular volume leads to a reduction in ventricular preload, which in turn results in a diminished stroke volume. Compensation occurs to a degree with an increase in HR. However, shock also causes a reduction in myocardial compliance, resulting in a decreased ventricular end-diastolic volume and thus stroke volume at any given ventricular filling pressure.
Neurohumoral Response
Sustained compensatory mechanisms exist to restore intravascular volume, maintain central perfusion, and mobilize metabolic and osmotic substrates. Disinhibition of the vasomotor center results in increased adrenergic output and decreased vagal activity. Decreased renal blood flow leads to renin release. Renin induces the formation of angiotensin I, which is then converted to angiotensin II. This is a potent vasoconstrictor and stimulates aldosterone release by the adrenal cortex. Aldosterone contributes to restoration of the intravascular volume by enhancing sodium reabsorption in the cortical collecting duct, in exchange for potassium and hydrogen. Hypovolemia, as well as an increase in serum osmolarity, leads to the release of antidiuretic hormone. This has a direct vasoconstrictive effect on vascular smooth muscle and also increases reabsorption of water in the distal renal tubule. The peripheral vasoconstriction is countered by circulating vasodilators including prostacyclin and nitric oxide and, more importantly, the products of local metabolism such as adenosine. This balance between the various vasoconstrictor and vasodilator effects determines local perfusion in a deleterious, repeating hypoxia–reperfusion cycle. Additionally, shock causes release of stress hormones: ephinephrine, adrenocorticotrophic hormone (ACTH), cortisol, and glucagon. This results in glycogenolysis, lipolysis, gluconeogenesis, and insulin resistance causing a negative nitrogen balance and a high extracellular concentration of glucose. This increase in glucose acts as a major osmotic agent for intravascular restitution and anaerobic energy supply. Insulin-independent tissues such as the brain and heart have a substantial increase in glucose utilization.
Pulmonary Response
The pulmonary vascular bed responds to shock much in the same way as the systemic vascular bed. The relative increase in pulmonary vascular resistance (PVR), particularly in septic shock, may exceed that of the SVR, potentially resulting in acute right heart failure. Shock results in tachypnea that reduces tidal volume and increases both dead space and minute ventilation, producing an early respiratory alkalosis. Hypoxia occurs due to diffuse alveolar damage, resulting in acute lung injury and subsequent acute respiratory distress syndrome (ARDS). These disorders are characterized by noncardiogenic pulmonary edema secondary to widespread pulmonary capillary and alveolar injury. Hypoxemia results from a significant ventilation–perfusion mismatch, and lung compliance is reduced due to loss of surfactant and lung volume, particularly functional residual capacity, in conjunction with an increase in intra-alveolar and interstitial edema.4,5
Renal Response
The kidneys respond to hypoperfusion by conserving sodium and water, with subsequent decreased urine output. Acutely, oliguria in the face of hypovolemia represents renal success, not renal failure. Early aggressive volume replacement has reduced the frequency of acute renal failure. Acute tubular necrosis is mostly due to the multifactorial interaction of shock, sepsis, nephrotoxic agents, and rhabdomyolysis. These toxic insults cause necrosis of tubular epithelium and tubular obstruction by cellular debris with back-leak of filtrate. Prolonged renal hypoperfusion results in a depletion of renal ATP stores with subsequent impairment of renal function and ultimate failure.
Inflammatory Response
The progression of shock is influenced by activation of the innate immunoinflammatory system. There is also a simultaneous anti-inflammatory response and suppression of the adaptive immune system in those surviving the initial insult. Excesses of both proinflammatory and anti-inflammatory responses are detrimental. An uncontrolled proinflammatory response results in a systemic inflammatory response syndrome, which may lead to continuing shock and MOF.6,7 Excessive anti-inflammatory responses and adaptive immune suppression increase the susceptibility to secondary nosocomial infections.
The complement cascade is activated by hypoxia and tissue injury, resulting in the generation of anaphylatoxins C3a and C5a. Direct complement fixation in injured tissues can progress to the C5-C9 attack complex, resulting in further cell damage. In addition, concomitant coagulation cascade activation causes microvascular thrombosis, with subsequent fibrinolysis and repeated episodes of ischemic/reperfusion injury. Thrombin is a potent inflammatory mediator that increases activation of neutrophils, leading to further microvascular injury.
The membrane Toll-like receptors are a major source of monocyte/macrophage activation. These receptors recognize damage-associated molecular patterns and pathogen-associated molecular patterns, which are released following tissue injury and pathogenic microbial organisms. Monocytes and macrophages are major regulators of the inflammatory response. Macrophages release proinflammatory cytokines such as interleukin 1 and 6 and tumor necrosis factor α (TNF-α). These produce many of the shock features including hypotension, lactic acidosis, and respiratory failure. Interleukin 8 is a potent neutrophil chemoattractant and up-regulates neutrophil adhesion molecules to enhance aggregation, adherence, and damage to the vascular endothelium.
Eicosanoids, such as prostaglandins, leukotrienes, and thromboxane A2, are vasoactive and immunomodulatory products that affect vascular resistance as well as capillary permeability. LTB4 attracts neutrophils and stimulates the formation of reactive oxygen species.
CLASSIFICATION OF SHOCK STATES
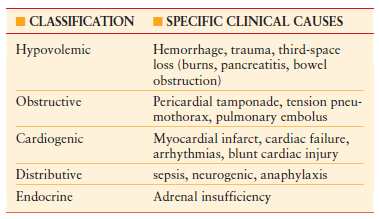
Although the cellular effects of shock are the common pathway regardless of the primary cause, a classification scheme is useful to delineate the underlying process. It is important to note that a combination of two or more causes of shock frequently occurs. The primary importance of classifying the type of shock lies in the recognition of the precipitating event, and initiating appropriate therapy to correct this original cause, while simultaneously carrying out resuscitation. There are five broad categories of shock as outlined in Table 4.1.
TABLE 4.1
CLASSIFICATION OF SHOCK STATES
GENERAL APPROACH TO SHOCK
The main priorities when dealing with shock include appropriate monitoring in an intensive care setting, prompt correction of tissue perfusion, and perhaps most importantly, correction or treatment of the precipitating cause. Careful and continuous assessment of physiological status and response to therapy is essential.
Physiologic Monitoring
Physiologic monitoring of the patient in shock commences with the “vital signs,” and progresses as needed, using increasing technology and invasiveness.
Heart rate is commonly elevated in response to intravascular volume loss, to maintain adequate CO. This tachycardia becomes pathologic when HR exceeds 120-130 beats per minute. Above this rate, there is insufficient diastolic time to adequately fill the ventricles, resulting in decreased stroke volume. A rapid decrease in HR after a volume challenge can be a useful indicator of hypovolemia. In contrast, bradycardia is associated with severe physiologic derangement, usually hypoxia and impending cardiovascular collapse. It is also seen in conjunction with neurogenic shock, as a result of disruption to the cardiac sympathetic fibers. Frequent use of beta-blockers to treat baseline cardiac disease also produces inappropriate HR responses.
Blood pressure (BP) is a crude measure of tissue perfusion. Hypotension results in inadequate tissue perfusion; however, the exact definition of hypotension is variable, depending on the individual patient. Elderly patients, in particular, are frequently hypertensive at baseline and may not be perfusing tissues adequately at a “normal” BP. Chronic use of antihypertensives also makes reliance on BP as a measurement of shock inaccurate. BP may be measured noninvasively or invasively. Both methods are subject to dynamic response artifacts that may result in inaccurate measurements. Obese patients frequently have unreliable BP measurements due to positioning and poor cuff compression. Mean arterial pressure (MAP) is usually consistent, regardless of measurement method and artifact, and is thus preferred.
Urine output is often decreased in shock in an attempt to preserve and restore intravascular volume. Oliguria is one of the earliest signs of inadequate tissue perfusion. Improvement of urine output is a key monitor for shock resuscitation.
Temperature may help define etiology of shock and provide prognostic value. Hypothermia results as an imbalance between heat loss and the body’s ability to generate and maintain metabolic energy. Clinically important hypothermia occurs when the core temperature is <35°C. Hypothermia is associated with an increase in sympathetic drive with resulting peripheral vasoconstriction, end-organ hypoperfusion, and metabolic acidosis from anaerobic respiration. In addition, hypothermia may exacerbate coagulopathy by causing dysfunction of the intrinsic and extrinsic coagulation pathways, as well as platelet activity. There is a markedly increased mortality in shocked patients with hypothermia.8,9
Pulse oximetry is a useful method of continuously monitoring arterial oxygen saturation. It utilizes the differential light absorption characteristics of oxyhemoglobin and deoxyhemoglobin to calculate the percentage saturation of oxygenated hemoglobin in blood. Pulse oximetry provides an indication of the degree of hypoxia as well as the assessment of oxygen transport balance.10
Biochemical markers are used to identify shock in early stages. The biochemical analysis of shock is based on the shift from aerobic to anaerobic metabolism with a resulting increase in lactate production and decreased clearance due to impaired hepatic function.11 Shock is also associated with a base deficit on arterial blood gas analysis. The base deficit is defined as the amount of fixed base or acid that must be added to an aliquot of blood to restore the pH to 7.40. Any patient with a lactate of ≥4 mmol/L or a base deficit of ≥6 mEq/L should be considered to be in shock until proven otherwise.12
Invasive Hemodynamic Monitoring
Most patients in the intensive care unit (ICU) may be managed safely without the use of a pulmonary artery catheter (PAC or Swan-Ganz catheter).13 However, in those patients with major ongoing blood loss, fluid shifts and/or underlying cardiac dysfunction, a PAC may be helpful. The current generation of PACs has been improved from the original catheter allowing measurement of pulmonary artery pressures, and continuous hemodynamic and oxygen transport assessment. The PAC is inserted percutaneously via the subclavian or internal jugular vein, to lie in the pulmonary artery.
The circulatory system consists of the systemic and pulmonary vasculature circuits connected in series. Two pressures are generated by each circuit: an outgoing pressure (MAP or mean pulmonary arterial pressure) and an incoming pressure or preload (pulmonary artery occlusion pressure [PAOP] or central venous pressure [CVP]), respectively. These pressures can be used to calculate the afterload of each circuit (SVR or PVR). Therefore, three variables are measured by the PAC: pressure, volume, and flow. From these measured variables, a number of calculated variables may be obtained, which can be useful in guiding resuscitative therapy. Table 4.2 outlines the hemodynamic variables obtained with a PAC.
TABLE 4.2
HEMODYNAMIC VARIABLES
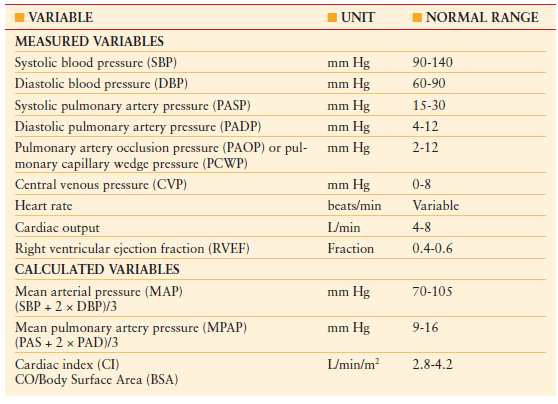
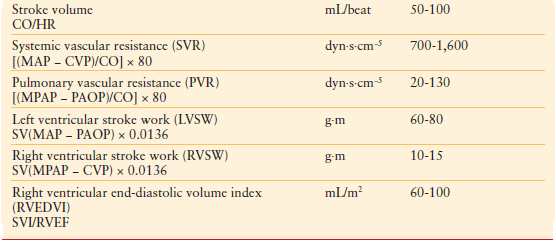
Preload optimization is essential in the initial resuscitation of all forms of shock. CVP and PAOP are commonly used to estimate preload. The Frank-Starling law defines preload in terms of myocardial fibril length at end diastole. As this is not measureable, several assumptions are made to utilize PAOP to assess the preload of the left ventricle. First, left ventricular end-diastolic volume is assumed to be proportional to myofibril length. Second, assuming that ventricular compliance is constant, end-diastolic volume is equal to end-diastolic pressure. Third, assuming that mitral valve function is normal, left ventricular end-diastolic pressure is equal to mean left atrial pressure (LAP). Fourth, properly transduced PAOP is equal to LAP. Similar assumptions are applicable to the use of CVP in estimating preload status of the right ventricle. While indirectly a surrogate for intravascular volume, overall these pressures correlate poorly with volumes.14 In addition, many of these assumptions are invalid in the critically ill patient due to changes in ventricular compliance, contractility, and intrathoracic pressures. Therefore, the trend (rather than absolute values of PAOP and CVP measurements) in response to therapeutic interventions is more useful and valid.
With the improvement in technology, current-generation PACs also allow a volumetric, as opposed to pressure-based, estimate of intravascular volume status. The right ventricular end-diastolic volume index (RVEDVI) is an accurate indicator of the effect of right ventricular preload on ventricular volume and thus output. Use of RVEDVI is significantly more accurate than PAOP or CVP in estimating cardiac preload and has been associated with a decrease in mortality and MOF when used in part as an end point of resuscitation.15–17
There is ongoing debate as to the utility and safety of PACs. A multicenter study in a mixed ICU population suggested that the use of a PAC was associated with an increased mortality.18 Also, several studies have suggested that there is no advantage to using a PAC versus a CVP monitor in the treatment of a mixed population of patients with adult respiratory distress syndrome.19 As a result, additional methods of volumetric preload assessment have been developed. Lithium dilution cardiac output (LiDCO) is a noninvasive method for measuring CO. It requires only an arterial pressure catheter and a central venous catheter. Lithium dilution is used to calibrate a pulse pressure algorithm, which allows continuous CO assessment.20 Although comparable with PACs, the LiDCO method does not allow continuous assessment of oxygen transport. The major problem is the loss of reliability in the acutely unstable patient.
In addition to measurements of CO, indicators of preload responsiveness including systolic and pulse pressure variation (SPV and PPV) can be obtained. These variations have been derived from observations in mechanically ventilated patients, where cyclical changes due to respiration are induced in the vena cava, pulmonary artery, and aortic blood flow. It is important to note that these arterial pressure variations are not an indicator of volume status, nor a direct marker of cardiac preload. Rather, SPV and PPV are indicators of the position on the Frank-Starling curve. Patients on the flat portion of the curve are insensitive to cyclical changes in preload induced by mechanical ventilation. Conversely, arterial pressure variations are increased in patients who are on the steep portion of the preload curve.21–23 Thus, a PPV of >12% or SPV >5 mm Hg is indicative of preload responsiveness and need for increased intravascular volume.20–23 The variables obtained by this method can be affected by aortic regurgitation, cardiac arrhythmia including tachycardia, irregular breathing, damped arterial line, or marked peripheral arterial vasoconstriction.
Esophageal Doppler ultrasound and transesophageal echocardiography have also been utilized in the hemodynamic assessment and monitoring of critically ill patients.24 These modalities provide information about global and ventricular function of the heart. End-diastolic volume may be assessed and has been shown to be more accurate than PAOP in the estimate of optimal preload status. Again, while these methods are comparable to several measurements obtained with PAC use, they do not allow continuous assessment of cardiopulmonary function and provide only a “snapshot” view of a complex, potentially rapidly changing physiology. In addition, these methods suffer the common problem of noninvasive monitors in that the algorithms and validation studies have been performed in relatively healthy patients. Algorithms used to calculate CO and stroke volume may be flawed in critically ill patients with severe shock.
Invasive Oxygen Transport Monitoring
In addition to the hemodynamic variables obtained with the PAC, determination of oxygen content in arterial and venous blood, together with CO and hemoglobin concentration, allows calculation of oxygen delivery, oxygen consumption, and oxygen-extraction ratio. The shock state induces an imbalance between oxygen demand and oxygen supply, resulting in anaerobic metabolism, lactic acidosis, and cell death. Table 4.3 outlines the oxygenation variables that may be obtained.
TABLE 4.3
OXYGEN TRANSPORT VARIABLES
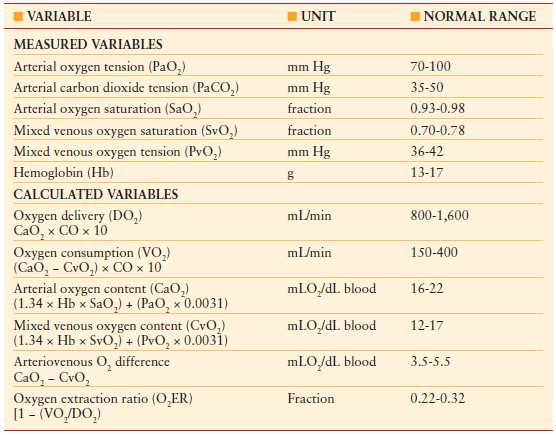
The assessment of oxygen transport begins with the calculation of oxygen content in blood. The majority of oxygen is bound to hemoglobin (>98%). Less than 2% is dissolved in plasma due to oxygen’s low solubility coefficient. Delivery of oxygen to the tissues is highly dependent upon the hemoglobin concentration as well as the ability of red blood cells to unload oxygen, which is dependent on the 2,3-diphosphoglycerate (2,3 DPG) content and its loss during blood banking storage. Oxygen delivery (DO2) and consumption (VO2) may be calculated by utilizing cardiac index (CI), arterial oxygen content (CaO2), and mixed venous oxygen content (CvO2).
The oxygen content in the pulmonary end capillary is at its highest, as none of the oxygen has been consumed by the tissues or diluted by unsaturated blood. As the blood leaves the heart, the oxygen content is reduced due to introduction of unsaturated blood from three sources. The first is bronchial blood, which empties into the pulmonary veins. The second is intrapulmonary shunt, which may be significantly higher in those patients with pulmonary dysfunction or in shock. The third is venous blood from the thebesian veins, which drain directly into the left ventricle after supplying the myocardium. Therefore the arterial O2 concentration can be calculated as the amount of oxygen bound to arterial hemoglobin + oxygen dissolved in arterial plasma.
After extraction of oxygen by the tissues, the blood is returned to the heart. The partial pressure of mixed venous oxygen tension (PvO2) can be measured by a venous blood gas or via the intra-atrial port of the PAC. The venous oxygen content of blood as it returns to the heart is calculated as the amount of oxygen bound to venous hemoglobin + oxygen dissolved in venous plasma.
The difference between arterial and venous oxygen content (CaO2 – CvO2) represents the amount of oxygen extracted by the tissues. It is often elevated in shock due to increased oxygen demands and decreased oxygen delivery. A CaO2 – CvO2 of >5.5 mL O2/dL or >35% suggests that CO is inadequate to optimally meet cellular oxygen demands.25 Ideally, adequate resuscitation should generate a mixed oxygen saturation of >0.70.
As seen in the calculation for oxygen delivery, an important factor is hemoglobin concentration. Previously, it was thought that a hemoglobin concentration of 10–13 g/dL was optimal for oxygen delivery; however, recent studies have suggested that transfusion to this level provides no benefit in critically ill patients and is associated with a decrease in survival. A hemoglobin concentration of <7–8 g/dL is recommended as the level required to trigger a blood transfusion, in the absence of recent acute myocardial infarct, unstable angina, or ongoing blood loss.26 Blood transfusion to maximize oxygen delivery must be balanced against the potentially harmful effects and increased risks of infection, immunosuppression, and organ failure associated with transfusion of banked blood.
GENERAL SHOCK TREATMENT PRINCIPLES
There are three priorities in the treatment of shock. First, the diagnosis and underlying cause of shock must be diagnosed and corrected. Second, resuscitation for shock must rapidly restore tissue perfusion and optimize oxygen delivery, hemodynamics, and cardiac function. Third, end-organ failure must be prevented or function supported. Often, resuscitation will be initiated prior to or simultaneously with identifying the underlying etiology.
For resuscitation, a reasonable goal of therapy is to achieve normal mixed venous oxygen saturation and arteriovenous oxygen-extraction ratio, while simultaneously, the elevated SVR should return to normal. Oxygen delivery may be enhanced by improving hemoglobin concentration, arterial oxygen saturation, and CO, individually or simultaneously.27 An algorithm for the resuscitation of the shocked patient is shown in Algorithm 4.1.
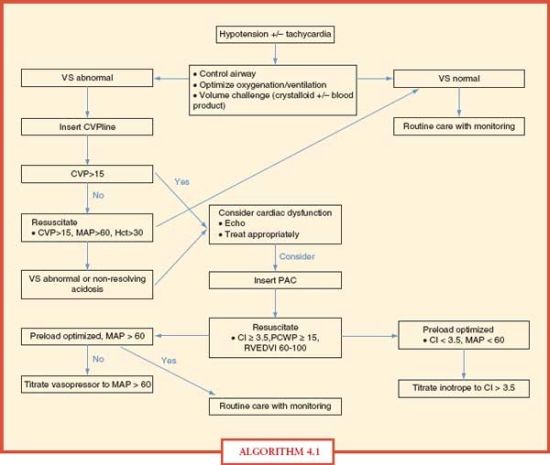
ALGORITHM 4.1 An algorithm for shock resuscitation. (VS, vital signs; CVP, central venous pressure; HCt, hematocrit; ECHO, echocardiogram; PAC, pulmonary artery catheter; CI, cardiac index in (L per minute) per m2; PCWP, pulmonary capillary wedge pressure in mm Hg; RVEDVI, right ventricular end-diastolic volume index.)
Fluid Therapy for Shock
A decreased CVP and oxygen delivery were key features noted in the early investigations of hemorrhagic shock, and a prolonged decrease in oxygen delivery led to a reduction in oxygen consumption. After successful fluid resuscitation, oxygen delivery and consumption increased above baseline for several hours. Failure of the patient to achieve this adequate response to resuscitation was almost universally fatal. Thus, restoration of hemodynamics and oxygen transport with fluid and inotropes became the primary paradigm of shock treatment. However, it is important to note that excessive use of either modality has been shown to be detrimental to patient outcomes and survival.
It was noted by Shires et al. that additional fluid replacement beyond the actual amount of blood loss, due to ongoing interstitial “third spacing” loss, was required to improve outcomes in hemorrhagic shock.28,29 These data led to the appropriate practice of aggressive, early fluid resuscitation. However, over time, complications from excessive fluid therapy began to appear, such as ARDS, abdominal compartment syndrome, and detrimental elevations in intracranial pressures. Currently, resuscitation volumes have become more judicious, and earlier use of blood components is advocated.
Crystalloids are balanced salt solutions widely used as resuscitative fluids. They restore the extracellular volume, decreasing the transfusion requirement after hemorrhagic shock and restoring adequate volume in most cases. Crystalloids are inexpensive and readily available. Lactated Ringer’s solution is isotonic and rapidly replaces the loss of interstitial fluid, without worsening any pre-existing electrolyte abnormality. However, recent investigations have identified the proinflammatory effects of lactated Ringer’s solution containing the D(-) isomer of lactate found in some commercial forms of lactated Ringer’s. D(-) lactate has been shown to increase neutrophil adhesion and increase production of reactive oxygen species.30,31 These adverse effects of crystalloid resuscitation are not found with L(-) lactate isomer formulations. In addition, lactated Ringer’s contains potassium and must be used cautiously if renal function is impaired or unknown.32 Normal saline solution is also effective in the resuscitation of shocked patients. Large-volume administration of normal saline, though, is associated with a hypernatremic, hyperchloremic metabolic acidosis. This acidosis complicates the resuscitation of the critically ill patient.
Colloids theoretically remain in the intravascular space for a longer period compared with crystalloids, due to the increased molecular size. Commonly used colloids include albumin, modified fluid gelatin, dextran 70, dextran 40, and hydroxyethyl starch. Despite widespread use since the early 20th century, there has been no demonstrated benefit to colloid use.33 In fact, colloid albumin infusion has been shown to increase mortality as well as prolong the resuscitation phase and delay postresuscitation diuresis. Albumin may also depress circulating immunoglobulin levels and reduce endogenous albumin synthesis. Given the lack of benefit and increased cost, colloids cannot be recommended in the current management of shock.34
Pharmacological Support
Pharmacological agents affecting preload, cardiac contractility, and afterload can be of major benefit in the treatment of shock. Importantly, optimal intravascular restoration must always precede cardiac augmentation. Inotropic support may be required when adequate preload fails to provide sufficient CO to meet tissue oxygen demands.35 Pressor support may be required when the systemic vasculature is abnormally dilated with a resulting diminished SVR, as seen particularly with hyperdynamic septic shock. The effect of inotropes and pressors is dependent on the specific adrenergic receptor affinity (Table 4.4). In cases of primary cardiac dysfunction, vasodilators can reduce cardiac oxygen demand by reducing afterload and/or by dilating the venous system and reducing preload. Afterload reduction may preserve stroke volume in the failing myocardium, whereas preload reduction may relieve pressure-driven pulmonary edema.
TABLE 4.4
HEMODYNAMIC EFFECTS OF INOTROPES/PRESSORS
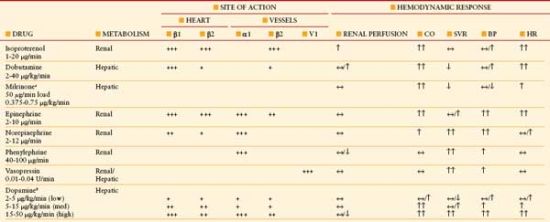
α1, vasoconstriction peripheral arterioles; β1, myocardial inotropy, chronotropy, enhanced AV conduction; β2, chronotropy, vasodilation mesenteric/skeletal bed, bronchodilation; V1 vasoconstriction peripheral arterioles, CO, cardiac output; SVR, systemic vascular resistance; BP, blood pressure; HR, heart rate.
aMilrinone: inhibits phosphodiesterase 3 resulting in increase in cAMP.
bDopamine: dopamine receptor stimulation at low dose vasodilates mesenteric/coronary/renal beds.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


