Thomas K. Henthorn
This chapter will review the fundamentals of clinical pharmacology to enable the anesthesia practitioner to use the information in subsequent chapters to develop anesthetic and postoperative analgesic plans that minimize exposure of patients to supratherapeutic and subtherapeutic concentrations and decrease patient morbidity. Opioids are used as a prototype drug class to explain these pharmacokinetic and pharmacodynamic principles.
I. Pharmacokinetics
In order to produce a drug effect, it is necessary to deliver the drug to the site of action. Most drugs used outside the perioperative or intensive care environment are administered in a manner in which their effects are achieved over days, weeks, or even years. In contrast, in the acute care environment, the clinician often needs to achieve onset of drug effect in a matter of minutes. Mathematical models that describe the drug plasma concentration versus time profile are readily available and easily implemented on model-based infusion pumps, personal computers, and even mobile devices. This initial section will qualitatively describe pharmacokinetic concepts that are necessary in order to rationally choose dosing regimens for opioids, hypnotics, and neuromuscular junction blocking agents.
A. Routes of Drug Administration
In order to produce a drug effect, it is necessary to deliver the drug to the site of action. Although the most familiar routes of drug administration are the oral and intravenous (IV) routes, there are a wide range of other sites for administering drugs into the body (Table 7-1). Blood flow is the usual means by which drug is delivered to the site of action. Thus, IV drug administration is considered the gold standard to which all other methods of drug administration are compared (i.e., bioavailability is defined as the ratio of drug exposure of a dose delivered by another route vs. IV administration). The IV route is preferred for several fundamental reasons. First, the IV route of drug administration provides almost immediate changes in drug concentrations in blood because the drug does not need to be absorbed into the systemic circulation. Furthermore, the vascular system is able to immediately deliver drugs to the site of drug action by intravascular mass transport to tissue capillary beds. Once drug reaches the capillary, it can enter tissue by diffusion or active transport. If extensive alveolar endothelial, epithelial drug uptake, or metabolism is present for inhalation anesthetics and vaporized drugs, bioavailability will be limited. Finally, the intravascular space is the easiest and most relevant biologic fluid to sample for drug concentration measurements. When multiple blood samples are obtained, the resultant drug concentration versus time data can be mathematically modeled to describe and interpret the pharmacokinetic properties of the drug. When injected or rapidly infused, IV drug administration results in rapid increases in drug–blood concentration, rapid transfer to tissues of the effect site, and rapid onset of drug effect. In general, the maximum drug effect is proportional to the maximum plasma concentration achieved.

Blood is rarely the site at which a drug produces its desired and undesired actions.
Table 7-1 Routes of Drug Administration Using Opioids as a Prototype
Routes of drug administration such as oral, intramuscular, and subcutaneous usually result in a slower onset of drug effect and a lower peak drug effect (Table 7-1). The rate and extent of drug absorption into the systemic circulation are important determinants of the time course of drug effect. The rate of drug absorption is dependent on a complex interplay of the physiochemical properties of the drug, the physiochemical properties of the delivery vehicle, and the perfusion of the tissue where the drug is administered.

For anesthetic gases and inhaled vaporized drugs, the bioavailability is close to 100%. Thus, the inhaled route mimics IV administration.
A few generalizations can be made regarding drug absorption. First, lipophilic drugs are absorbed faster than hydrophilic drugs because diffusion across lipid cellular barriers is faster. Furthermore, drug delivery vehicles or devices can be designed that slow the rate of drug absorption by such mechanisms as physically binding a drug molecule to decrease its lipophilicity (e.g., sustained-release drug formulations of pills) or adding additional diffusion barriers (e.g., transdermal patch layers). Finally, the perfusion of the tissue where the drug is administered can significantly alter its absorption profile. For example, inhaled fentanyl via a microvaporization device has a plasma concentration profile that is similar to that of IV drug administration. This is because nearly all the drug reaches the alveoli, where there is a minimal diffusion barrier and perfusion by the entire cardiac output. In contrast, oral administration of drugs produces a peak plasma concentration that is substantially lower than IV administration. There is a delayed time to peak concentration because of the limited perfusion of the gastrointestinal mucosa and the metabolism of drug in the liver before entering the systemic circulation, thus limiting both the rate and extent of absorption, respectively. This first-pass metabolism by the liver is the major determinant of the limited bioavailability of many drugs administered via the gastrointestinal tract. For neuraxial routes, the physiochemical properties of the drug and the physiochemical delivery of the vehicle determine the rate by which drug diffuses locally into adjacent neural tissue. Uniquely, systemic absorption and the plasma drug concentration profile are not related to the drug effect profile by these routes.

There are two non-IV routes for which tissue perfusion does not affect drug onset of effect because the drug is delivered adjacent to the site of action—the intrathecal space and the epidural space.
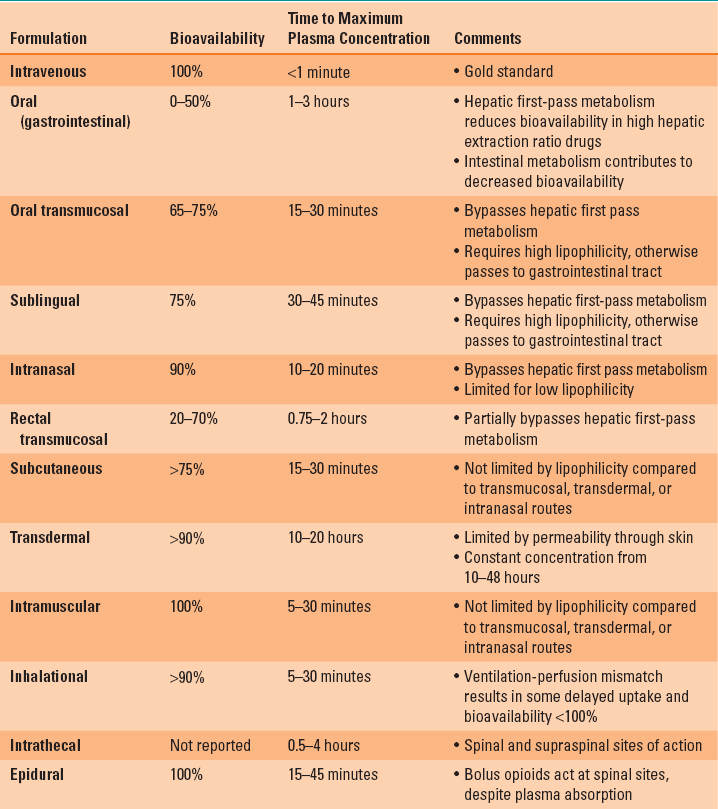
The slower rate of systemic absorption and the lower peak plasma concentration of the non-IV methods of drug administration may lead clinicians to believe that the only benefit of this route of drug administration is convenience in avoiding IV access. However, a commonly overlooked benefit of the non-IV methods of drug administration is that the same impediments to rapid systemic absorption can provide a prolonged drug effect. As long as the plasma concentration is maintained at a level above the therapeutic threshold concentration, the drug will produce an effect (Fig. 7-1) (1).
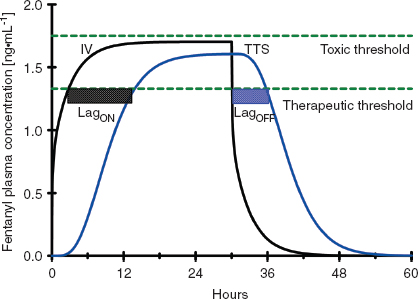
FIGURE 7-1 The plasma concentration profile achieved with 75 μg/hr of transdermal therapeutic systems fentanyl (blue line, TTS-fentanyl) or intravenous fentanyl (black line, IV-fentanyl). The TTS-fentanyl “simulation” is based on a compartmental model developed to mimic the output of published observations, whereas the IV-fentanyl simulation was generated from the most robust three-compartment fentanyl pharmacokinetic-pharmacodynamic (PK-PD) model available. Although there is a brief delay until the IV-fentanyl achieves a plasma concentration above the therapeutic threshold (lower dashed green line), the delay until the TTS-fentanyl produces a plasma concentration above the therapeutic threshold is much longer (black crosshatched bar, LagON). This delay results from a combination of the time that it takes the TTS-fentanyl system to establish and equilibrate with a subcutaneous depot of fentanyl and the continuous tissue distribution and systemic elimination of intravenously absorbed fentanyl. Similarly, when fentanyl administration is discontinued 30 hours after initiation of therapy, there is a rapid decrease in the plasma concentration in the IV-fentanyl profile, whereas the continued uptake of fentanyl from the subcutaneous depot maintains the plasma concentration above the therapeutic threshold for several hours in the TTS-fentanyl profile and then slows the rate at which the plasma concentration decreases. This delay (blue crosshatched bar, LagOFF) is not due to an alteration in the plasma distribution or the systemic elimination of fentanyl, but rather to the continued absorption from the subcutaneous depot.
B. Drug Distribution and Drug Elimination
Drug Distribution
Once drug is in the central circulation, it travels by intravascular blood flow to the tissue capillary beds where it is free to leave and re-enter the intravascular space by diffusion or active transport at the blood–tissue interface. Drug transport, either into or from the tissue, is usually not saturable, so drug uptake by the tissue is mostly limited by the blood flow to the tissue (flow-limited drug uptake). The distribution of cardiac output to the different tissue beds and the cellular mass of the tissue determine the rate of drug equilibration with the plasma in each tissue bed. For example, the brain and the kidneys equilibrate over a matter of minutes due to their high blood flow and relatively low tissue volume (2). In contrast, the well-perfused muscle and splanchnic tissues take hours to approach equilibrium due to their large tissue volumes relative to blood flow (Fig. 7-2). Despite having the highest blood to tissue solubility ratio, the body’s adipose tissue is relatively poorly perfused and therefore does not approach equilibrium with the plasma for days.
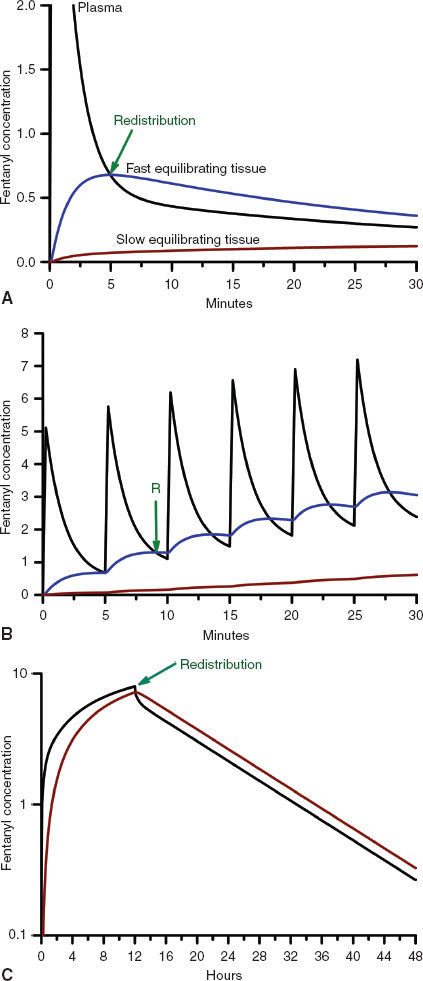
FIGURE 7-2 A: The concentration profiles produced by an intravenous bolus of fentanyl for the concentration in the plasma (solid black line), fast equilibrating tissue (solid blue line), and slow equilibrating tissue (solid red line). Note that the plasma concentration of fentanyl decreases rapidly after administration while it increases rapidly in the fast equilibrating tissue. From the point when the concentration in the fast equilibrating tissue exceeds that in the plasma (green arrow), there will be continued transfer of drug from the fast equilibrating tissue to the plasma (redistribution). Throughout this time there is continuous, slow uptake of drug by the slow equilibrating tissue and there is no redistribution of drug from this tissue to the plasma. B: The concentration profiles produced by repeated intravenous boluses of fentanyl every 5 minutes for the concentration in the plasma (solid black line), fast equilibrating tissue (solid blue line), and slow equilibrating tissue (solid red line). Note that the plasma concentration of fentanyl rapidly decreases after each bolus, while it rapidly increases throughout the entire 30-minute period in fast equilibrating tissue. For each bolus from the second bolus onward, there is redistribution of drug from the fast equilibrating tissue to the plasma at any of the times when the plasma concentration is lower than the fast equilibrating tissue concentration (initial time of redistribution is at the green arrow, R). With each subsequent bolus, the duration of redistribution is longer and longer. There is still no redistribution between the slow equilibrating tissue and the plasma. C: The concentration profiles produced by a 12-hour continuous infusion of fentanyl for the concentration in the plasma (solid black line) and slow equilibrating tissue (solid red line). Note that the fast equilibrating tissue is not shown because on this time scale it essentially overlaps that of the plasma, although, in reality there is a small difference that results in transfer of drug from the plasma to the fast equilibrating tissue until the infusion is stopped, at which time there is redistribution of drug from the fast equilibrating tissue to the plasma. Note that once the infusion is stopped, there is redistribution of drug from the slow equilibrating tissue to the plasma (green arrow). In addition, the elimination half-life is related to the slope of the washout curve (after the infusion is terminated).
Unless the tissue metabolizes or excretes the drug, drug transport between the blood and the tissue is a bidirectional process that is governed by the law of mass action. When the concentration of drug in the tissue becomes higher than the plasma concentration, the overall movement of drug is from the tissue to the plasma (Fig. 7-2). This redistribution of an opioid or hypnotic from the highly perfused but low tissue volume brain back into the plasma will eventually decrease the concentration of drug at its site of action below the therapeutic threshold and thereby halt its effect. It is important to note that the brain may redistribute drug minutes after a bolus of drug has been administered. But tissue beds such as the muscle, which are slower to equilibrate because of the large capacity for drug uptake, will continue to take up drug until the plasma concentration decreases below the tissue concentration as drug is metabolized, excreted, and drug distributed throughout the body. With repeated injections or an infusion of drug, tissue uptake of drug will continue as long as the blood–drug concentration remains above that of tissue–drug concentration. Thus, when drug administration is stopped or the rate is decreased, these tissues begin to act like depots, causing net transfer of drug to blood and slowing the rate of further decrease of the plasma concentrations (Fig. 7-2). When the rate of net tissue transfer of drug to blood is equal to the elimination rate of drug from the body, then the blood–drug concentration versus time profile enters into the terminal elimination phase where the rate of change (or half-life) remains constant (3).
Drug Elimination
The only way to decrease the total amount of drug in the body is to eliminate it from the body. Some drugs are sufficiently hydrophilic to be excreted via passive filtration or active transport unchanged by the kidneys. But the majority of opioids and hypnotics are sufficiently lipophilic (they must cross the blood–brain barrier) that they must be metabolized to a more hydrophilic form that can be excreted. Biotransformation of drugs into more hydrophilic compounds is usually achieved by enzymatic reactions that are oxidative or, rarely, reductive. Together these are referred to as phase I reactions that occur in the endoplasmic reticulum of liver cells, but also in some cases the kidney, intestinal mucosa, and lung. The cytochrome P450 (CYP P450) superfamily of enzymes, which are found in the endoplasmic reticulum of hepatocytes and some other cells, is primarily responsible for phase I reactions. These new molecularly altered drugs can then be conjugated to such molecules as glucuronide by enzymes, in this case, glucuronidase. Some drugs, such as propofol and morphine can be directly conjugated to glucuronide without phase I reactions. Regardless of whether there was a phase I step, conjugations are referred to as phase II reactions and occur in the cytosol of hepatocytes (and other cells). Glucuronides and other conjugates can then be eliminated into the urine or gastrointestinal tract via the bile.

Genetic polymorphisms of the enzymes involved in drug metabolism are the most common cause of interindividual variability in the rate of drug elimination from the body.
C. Mathematical Concepts in Pharmacokinetics
Volume of Distribution
In a purely physical sense, the amount (mass) of solute divided by a known physical volume of fluid will equal the concentration of the solute in the fluid. In a patient, this is strictly true as well, but one would have to homogenize the patient in order to measure the correct concentration in the resultant fluid volume. In pharmacokinetics, the sample fluid is typically plasma and the amount of solute (or drug) put into the body can also be measured. So the plasma-apparent volume of distribution becomes the drug (or solute) dose divided by the plasma drug concentration. If one could homogenize the patient, the volume of fluid in the patient would always be the volume of distribution of the solute (or drug). Almost always, what is reported in pharmacokinetic tables is the plasma-apparent volume of distribution. It rarely corresponds to any known true physical fluid volume and, instead, is what the volume appears to be based on the plasma–drug concentration (2,3).
Volumes of distribution of drugs can range from a practical low of the body’s extracellular fluid volume (for drugs that are excluded from entering cells) to volumes that are many times larger than the weight of a patient (for drugs that partition into tissues because of active transport, binding, or physicochemical preference for the lipid constituents of cells over the watery environment of plasma).
Elimination Clearance
As drug is delivered to excretory organs such as the liver or kidneys, the drug is subject to permanent removal or biotransformation to another chemical entity (i.e., it ceases to exist as the drug administered). If an organ perfectly removed all drug from the blood as it flowed through, the clearance would equal the blood flow to that organ. If only a fraction of the drug present in the blood to the organ can be removed in the time of passage dictated by blood flow, then clearance is the fraction removed on one pass multiplied by the organ’s blood flow. Thus, clearance is stated in terms of flow (e.g., liters per minute), and elimination clearance represents the total of all the clearances of all the eliminating organs for a particular drug.
Usually, drugs that have clearances near that of liver blood flow are very efficiently biotransformed by phase I reactions (e.g., fentanyl) or phase II reactions (e.g., propofol) in the liver and, in the case of propofol, also in the kidneys. With such drugs, the elimination clearance is considered to be flow limited,
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


