Paul S. Pagel
This chapter reviews the cardiovascular pharmacology of medications used to alter hemodynamics during surgery and in the intensive care unit, including endogenous and synthetic catecholamines, sympathomimetics, milrinone, vasopressin, and antihypertensive medications.
I. Catecholamines
The α, β, and dopamine adrenergic receptor subtypes are responsible for mediating the cardiovascular effects of endogenous (epinephrine, norepinephrine, dopamine) and synthetic (dobutamine, isoproterenol) catecholamines (Fig. 13-1). These drugs all activate β1 adrenoceptors located on the sarcolemmal membrane of atrial and ventricular myocytes to varying degrees. This β1 adrenoceptor stimulation causes positive chronotropic (heart rate), dromotropic (conduction velocity), inotropic (contractility), and lusitropic (relaxation) effects. The β1 adrenoceptor is coupled to a stimulatory guanine nucleotide-binding (Gs) protein that activates the key intracellular enzyme adenylyl cyclase, thereby accelerating the formation of the second messenger cyclic adenosine monophosphate (cAMP) from adenosine triphosphate (ATP) (Fig. 13-2). Three major consequences result from activation of this signaling cascade: (a) more calcium (Ca2+) is available for contractile activation; (b) the efficacy of activator Ca2+ at troponin C of the contractile apparatus is enhanced; and (c) removal of Ca2+ from the contractile apparatus and the sarcoplasm after contraction is accelerated. It should be readily apparent that the first two of these actions produce a direct increase in contractility, whereas the third results in more rapid myocardial relaxation during early diastole. Indeed, treatment of acute or chronic left ventricular (LV) dysfunction is the primary reason for the perioperative use of catecholamines. Notably, how well catecholamines work under these clinical conditions may be affected by the relative density and functional integrity of the β1 adrenoceptor and its signaling cascade because receptor down-regulation and abnormal intracellular Ca2+ homeostasis are characteristic features of heart failure.

Catecholamines activate β1 adrenoceptors, which in turn accelerate the formation of the second messenger, cyclic adenosine monophosphate.
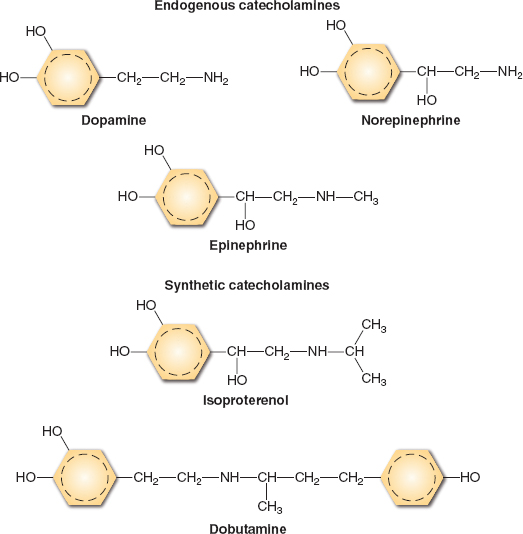
FIGURE 13-1 Catecholamine chemical structures.
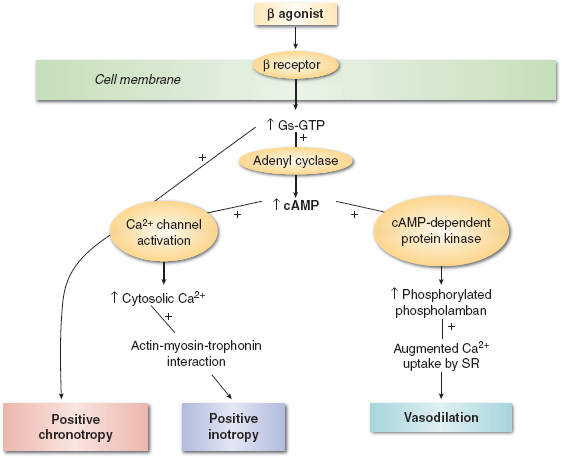
FIGURE 13-2 Schematic of β adrenergic agonist mechanism of action. (From Gillies M, Bellomo R, Doolan L, et al. Bench-to-bedside review: Inotropic drug therapy after adult cardiac surgery—a systemic literature review. Crit Care. 2005;9:266–279, with permission.)
The circulatory effects of catecholamines in other perfusion territories are dependent on the tissue-specific distribution of α and β adrenoceptor subtypes (Table 13-1). Differences in each catecholamine’s chemical structure and its relative selectivity for adrenoceptors also influence the peripheral vascular actions of these medications. This selectivity is often dose related (Table 13-2); dopamine provides a useful pedagogical illustration of this principle. Low doses of this catecholamine predominantly stimulate dopamine subtypes 1 and 2 (DA1 and DA2, respectively) receptors, causing arterial vasodilation, but progressively larger doses sequentially activate β1 and α1 adrenoceptors, augmenting contractility and causing arterial vasoconstriction, respectively. The α1 adrenoceptors are major regulators of vasomotor tone as a result of their location in arteries, arterioles, and veins. Thus, catecholamines that exert substantial α1 adrenoceptor agonist activity (e.g., norepinephrine) increase systemic vascular resistance and reduce venous capacitance through arterial and venous vasoconstriction, respectively. The α1 adrenoceptor-mediated vasoconstriction occurs through phospholipase C-inositol 1,4,5-triphosphate, signaling through an inhibitory guanine nucleotide-binding (Gi) protein (Fig. 13-3). This cascade opens Ca2+ channels, releases Ca2+ from intracellular stores (sarcoplasmic reticulum and calmodulin), and activates several Ca2+-dependent protein kinases; the sum total of these events causes an increase in intracellular Ca2+ concentration and vascular smooth muscle cell contraction. Whereas α1 adrenoceptors are the major target for catecholamines in cutaneous blood vessels, β2 adrenoceptors predominate in skeletal muscle, and activation of this latter adrenoceptor subtype produces arteriolar vasodilation via adenylyl cyclase–mediated signaling.

The α1 adrenoreceptors are major regulators of vasomotor tone, including systemic vascular resistance and venous capacitance.
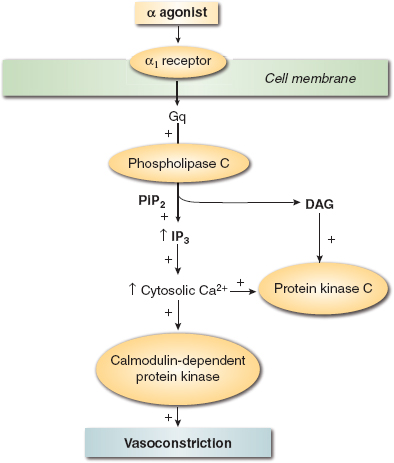
FIGURE 13-3 Schematic of α adrenergic agonist mechanism of action. (From Gillies M, Bellomo R, Doolan L, et al. Bench-to-bedside review: Inotropic drug therapy after adult cardiac surgery—a systemic literature review. Crit Care. 2005;9:266–279, with permission.)
Table 13-1 Adrenergic Receptors: Order of Potency of Agonists and Antagonists
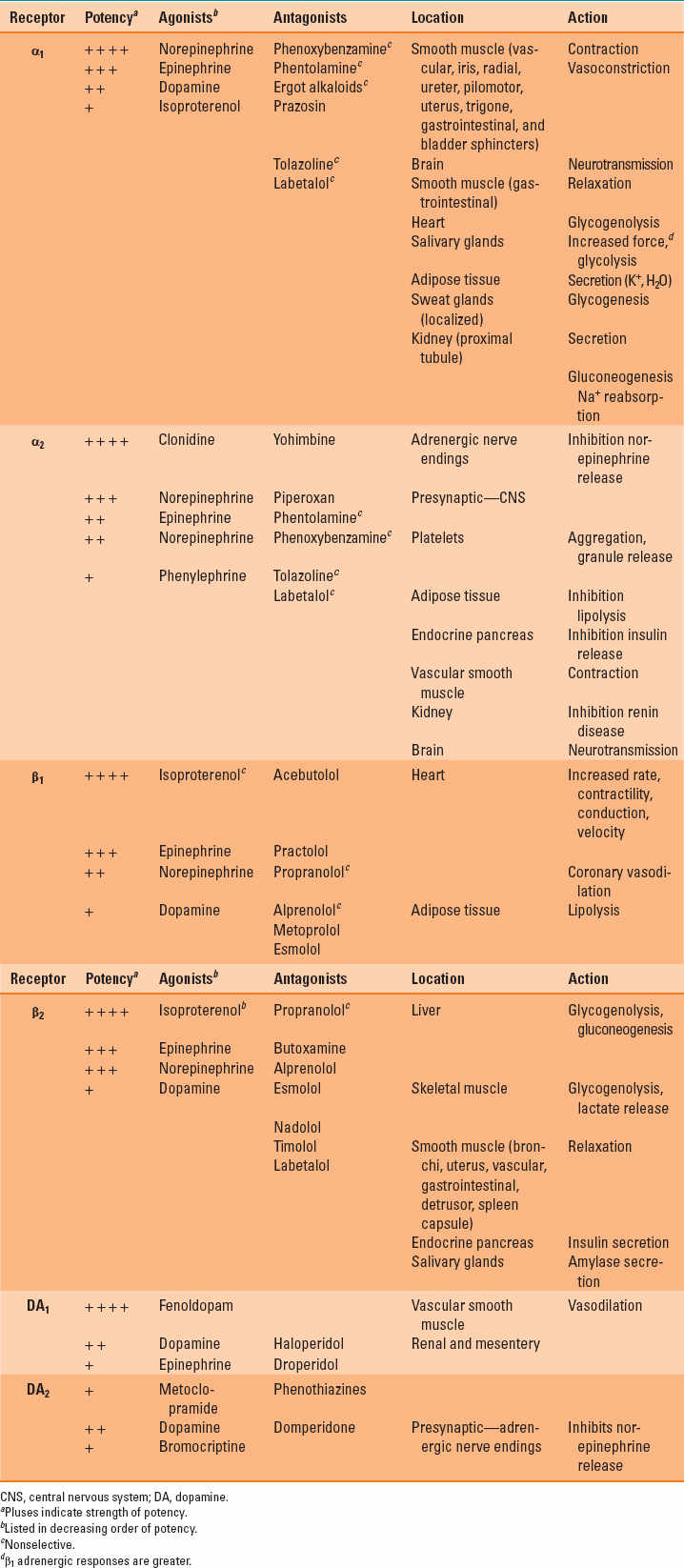
Table 13-2 Inotropic and Vasopressor Drug Names, Clinical Indication for Therapeutic Use, Standard Dose Range, Receptor Binding (Catecholamines), and Major Clinical Side Effects
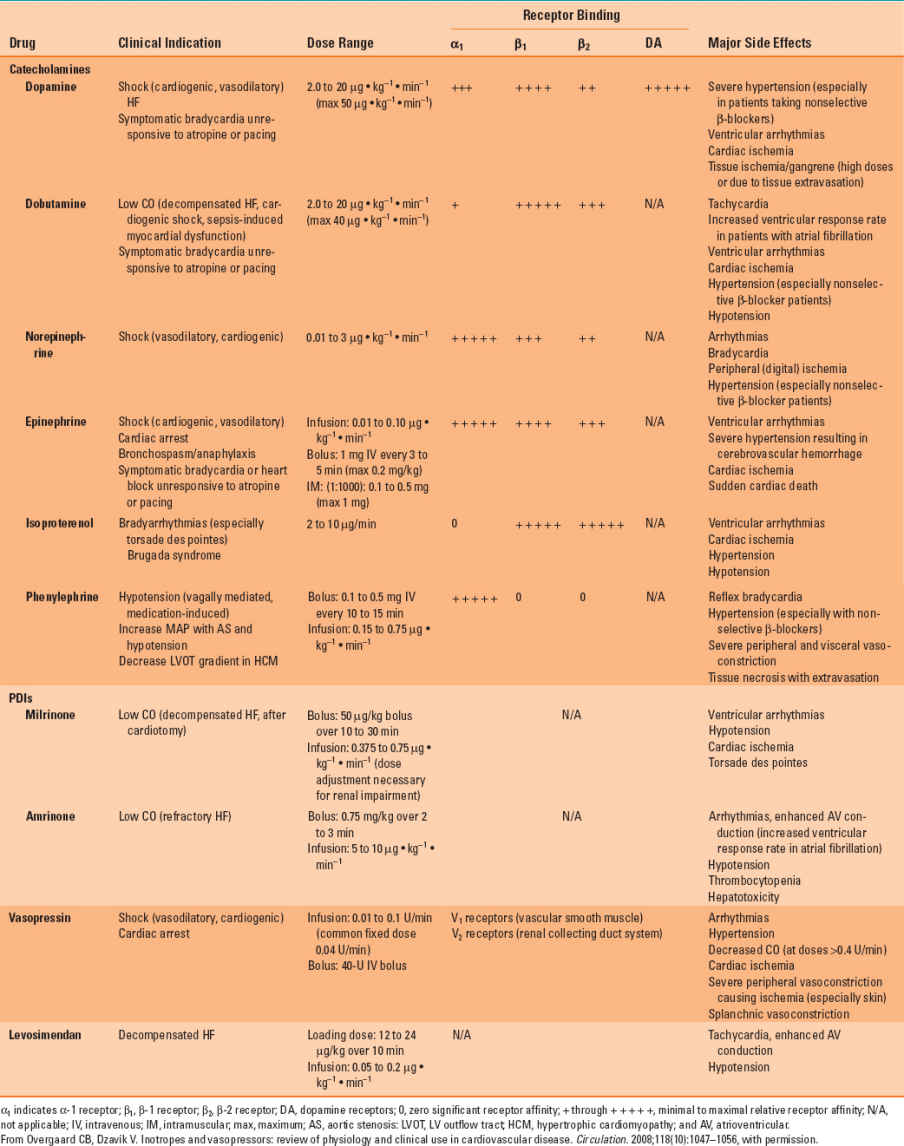
The actions of specific catecholamines on hemodynamics are summarized in Table 13-3. For example, if a catecholamine acts primarily through the α1 adrenoceptor (e.g., norepinephrine), an increase in arterial pressure will most likely be observed because enhanced arterial and venous vasomotor tone increases systemic vascular resistance (greater afterload) and augments venous return to the heart (increased preload), respectively. In contrast, a catecholamine with β1 and β2 adrenoceptor activity and little effect on the α1 adrenoceptor (e.g., isoproterenol) would be expected to modestly decrease arterial pressure because reductions in systemic vascular resistance offset increases in cardiac output caused by tachycardia and enhanced myocardial contractility. It is important to note that all catecholamines may cause detrimental increases in myocardial oxygen consumption in patients with flow-limiting coronary artery stenoses and, therefore, contribute to the development of acute myocardial ischemia. As a result, the use of catecholamines to support LV function in patients with coronary artery disease complicated by congestive heart failure requires substantial caution. Thus, it should come as no surprise that a drug that reduces LV afterload, and not one that causes a positive inotropic effect, is usually chosen first to improve cardiac output in a patient with coronary artery disease and heart failure.
Table 13-3 Dose Schedule and Hemodynamic Effects of the Adrenergic Agonists
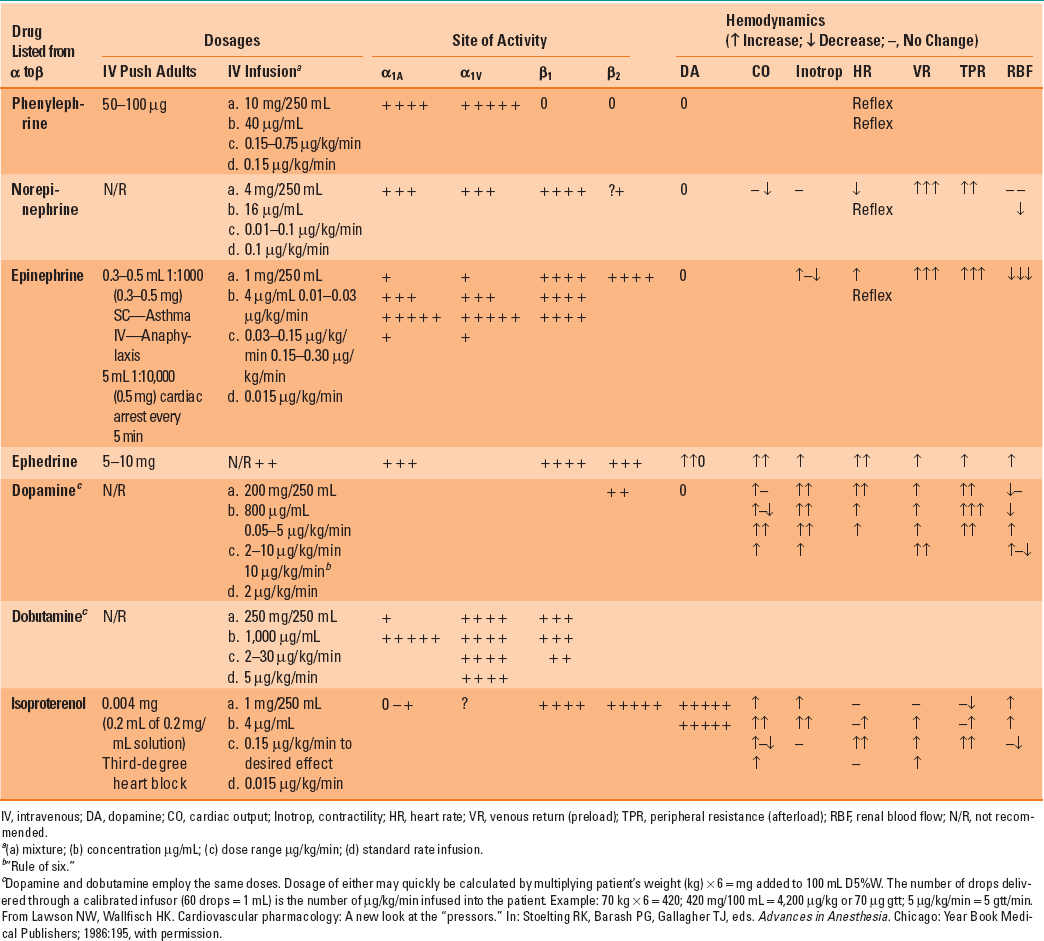
Table 13-4 Recommendations for Perioperative Beta-Blocker Therapy

A. Epinephrine
Epinephrine is an endogenous catecholamine that exerts its cardiovascular effects by activating α1, β1, and β2 adrenoceptors. Epinephrine stimulates β1 adrenoceptors located on the cell membranes of sinoatrial node cells and cardiac myocytes to produce positive chronotropic and inotropic effects, respectively. Epinephrine-induced activation of β1 adrenoceptors also enhances the rate and extent of myocardial relaxation, thereby facilitating greater LV filling during early diastole. The combination of these actions on heart rate and LV systolic and diastolic function causes pronounced increases in cardiac output. The tachycardia initially observed during an infusion of epinephrine may be subsequently attenuated as baroreceptor-mediated reflexes are activated. As a result, epinephrine is especially useful for the treatment of acute LV failure during cardiac surgery because it predictably increases cardiac output. Epinephrine also enhances cardiac output and oxygen delivery without causing deleterious increases in heart rate in septic, hypotensive patients.
It is important to note that clinical use of epinephrine may be limited because the catecholamine stimulates the development of atrial or ventricular arrhythmias. Epinephrine increases conduction velocity and reduces refractory period in the atrioventricular (AV) node, the His bundle, Purkinje fibers, and ventricular muscle. The positive dromotropic effect of epinephrine on AV nodal conduction may cause detrimental increases in ventricular rate in the presence of atrial flutter or fibrillation. Irritability in other parts of the conduction system may also precipitate ventricular arrhythmias, including premature ventricular contractions, ventricular tachycardia, and ventricular fibrillation, especially in the presence of a pre-existing arrhythmogenic substrate (e.g., regional myocardial ischemia or infarction, cardiomyopathy).
Epinephrine causes vasoconstriction of arteriolar vascular smooth muscle in the cutaneous, splanchnic, and renal perfusion territories through its effects at the α1 adrenoceptor, but the catecholamine also simultaneously produces vasodilation in the skeletal muscle circulation as a result of β2 adrenoceptor activation. These observations emphasize that the organ-specific distribution of α1 and β2 adrenoceptors determines epinephrine’s overall effect on blood flow. These effects are also dose dependent: lower doses of epinephrine stimulate β2 adrenoceptors, causing peripheral vasodilation and modest declines in arterial pressure, but higher doses of the catecholamine activate α1 adrenoceptors, thereby increasing systemic vascular resistance and arterial pressure. A high density of α1 adrenoceptors is also present in the venous circulation, and as a result, epinephrine produces venoconstriction and augments venous return. Epinephrine also causes vasoconstriction of the pulmonary arterial tree and increases pulmonary arterial pressures through α1 adrenoceptor activation. The α1 and β2 adrenoceptors exist in the coronary circulation, but these receptor subtypes play minor roles in establishing coronary perfusion during administration of epinephrine. Instead, epinephrine-induced increases in coronary blood flow occur principally because of metabolic autoregulation: increases in myocardial oxygen demand resulting from increases in heart rate, contractility, preload, and afterload are responsible for coronary vasodilation. Nevertheless, epinephrine may cause epicardial coronary vasoconstriction and reduce coronary blood flow in situations where maximal coronary vasodilation is present (e.g., acute myocardial ischemia distal to a severe coronary stenosis) via direct stimulation of α1 adrenoceptors.
Prior administration of α or β adrenoceptor antagonists influences the cardiovascular effects of epinephrine. For example, epinephrine causes greater increases in systemic vascular resistance and arterial pressure when administered after the nonselective beta-blocker propranolol because β2 adrenoceptor-mediated arterial vasodilation no longer opposes α1 adrenoceptor-induced vasoconstriction. Established beta-blockade also competitively inhibits β1 adrenoceptor activation by epinephrine, thereby attenuating the positive chronotropic and inotropic effects of the catecholamine. Such a competitive blockade may only be overcome by larger doses of epinephrine. Indeed, the hemodynamic effects of epinephrine may be similar to those the pure α1 adrenoceptor agonist phenylephrine (see below) in the presence of complete β1 and β2 adrenoceptor blockade.

In patients with complete β1 and β2 blockade, epinephrine produces hemodynamic effects similar to a pure α1 adrenoceptor agonist like phenylephrine.
B. Norepinephrine
Norepinephrine is the neurotransmitter that is released from sympathetic nervous system postganglionic neurons. This catecholamine activates both α1 and β1 adrenoceptors similar to epinephrine, but norepinephrine exerts few if any effects on the β2 adrenoceptor. As a result, norepinephrine enhances myocardial contractility while simultaneously causing arterial vasoconstriction. These actions dramatically increase arterial pressure, but cardiac output remains largely unchanged. In contrast, pure α1 adrenoceptor agonists such as phenylephrine cause predictable, dose-related decreases in cardiac output because simultaneous increases in contractility mediated through the β1 adrenoceptor do not occur. Unlike epinephrine, norepinephrine does not usually increase heart rate because elevated blood pressure stimulates baroreceptor-mediated reflexes. This action usually balances the direct positive chronotropism of β1 adrenoceptor activation. In general, greater increases in systemic vascular resistance and diastolic arterial pressure are observed during administration of norepinephrine compared with similar doses of epinephrine. Norepinephrine also causes constriction of venous capacitance vessels through α1 adrenoceptor stimulation, thereby increasing venous return and augmenting stroke volume.
Norepinephrine is especially useful for treatment of refractory hypotension during pathologic conditions in which pronounced vasodilation occurs. For example, norepinephrine increases arterial pressure, cardiac index, and urine output in patients with sepsis. Norepinephrine is also useful for the treatment of vasoplegic syndrome, a hypotensive state associated with low systemic vascular resistance that sometimes occurs during prolonged cardiopulmonary bypass in patients undergoing cardiac surgery. Norepinephrine increases coronary perfusion pressure in patients with severe coronary artery disease, but the drug may cause spasm of internal mammary or radial artery grafts used during coronary bypass artery graft (CABG) surgery through α1 adrenoceptor activation. Norepinephrine is associated with development of ventricular and supraventricular arrhythmias, but the potential arrhythmogenic effects of this catecholamine are less than those from epinephrine. As a result, it may be appropriate to substitute norepinephrine for epinephrine when treating cardiogenic shock in the presence of hemodynamically significant atrial or ventricular arrhythmias. Norepinephrine stimulates pulmonary arterial α1 adrenoceptors and causes dose-related increases in pulmonary arterial pressures that may precipitate right ventricular dysfunction because this chamber is less able to tolerate acute elevations in afterload than the LV. Addition of a selective pulmonary vasodilator such as inhaled nitric oxide may be beneficial to attenuate norepinephrine’s actions as a direct pulmonary vasoconstrictor when the drug is used to treat LV dysfunction in patients with pulmonary hypertension. Dose-dependent decreases in hepatic, skeletal muscle, splanchnic, and renal blood flow occur during administration of norepinephrine via α1 adrenoceptor activation when blood pressure is normal or modestly reduced. But in the presence of profound hypotension (e.g., sepsis), norepinephrine increases perfusion pressure and blood flow to these vascular beds. Nevertheless, sustained reductions in renal and splanchnic blood flow represent a major limitation of prolonged use of norepinephrine.
C. Dopamine
Dopamine is the biochemical precursor of norepinephrine and differentially activates several adrenergic and dopaminergic receptor subtypes in a dose-related manner. Low doses (typically below 3 μg/kg/min) of dopamine selectively increase renal and splanchnic blood flow via activation of DA1 receptors and also reduce norepinephrine release from autonomic nervous system ganglia and adrenergic neurons through a DA2 receptor–mediated mechanism. These combined effects produce a modest decline in arterial pressure. Moderate doses (3 to 8 μg/kg/min) of dopamine activate both α1 and β1 adrenoceptors, whereas high doses (in excess of 10 μg/kg/min) almost exclusively act on α1 adrenoceptors to increase arterial pressure through arteriolar vasoconstriction. However, it is important to realize that this straightforward dose–response description of dopamine pharmacodynamics is overly simplistic because differences in receptor density and regulation, drug interactions, and patient variability cause a broad range of clinical responses to the catecholamine. For example, low doses of dopamine were once thought to provide renal protection through DA1 receptor–mediated increases in renal blood flow alone. But it is now clear that even low doses of dopamine also stimulate α1 and β1 adrenoceptors, which may obfuscate the catecholamine’s intended dopaminergic effect. Conversely, renal blood flow and urine output may be maintained (and not decreased) during administration of higher doses of dopamine because DA1 receptors continue to be activated, despite a predominant α1 adrenoceptor agonist effect. Such varied responses may explain, at least in part, why dopamine fails to consistently provide renal protective effects, despite causing modest increases in renal perfusion and urine output.
Dopamine is often used for inotropic support in patients with acute LV dysfunction. This action occurs as a result of activation of β1 adrenoceptors. Dopamine also stimulates arterial and venous α1 adrenoceptors, thereby increasing LV afterload and enhancing venous return, respectively. Thus, dopamine augments arterial pressure. The use of dopamine for the treatment of hypotension associated with depressed contractile function may be limited to some degree in patients with pre-existing pulmonary hypertension or elevated preload. Right atrial, mean pulmonary arterial, and pulmonary capillary occlusion pressures were greater in patients undergoing cardiac surgery after cardiopulmonary bypass who had received dopamine compared with dobutamine, despite producing similar cardiac output. Dopamine may also cause greater increases in heart rate than epinephrine in cardiac surgery patients. Infusion of an arterial vasodilator (e.g., sodium nitroprusside) may be used to mitigate the increases in LV afterload associated with administration of dopamine and, in so doing, may further augment cardiac output. However, administration of an inotrope vasodilator (inodilator), such as milrinone, has largely replaced this “dopamine plus nitroprusside” approach. Like epinephrine and norepinephrine, dopamine increases myocardial oxygen consumption and may worsen myocardial ischemia in the presence of hemodynamically significant coronary stenoses.
D. Dobutamine
Dobutamine is a synthetic catecholamine composed of two stereoisomers (negative and positive), both of which stimulate β adrenoceptors, whereas these stereoisomers produce opposing agonist and antagonist effects on α1 adrenoceptors. As a result, dobutamine causes potent β adrenoceptor stimulation, but exerts little or no effect on α1 adrenoceptors when administered at infusion rates <5 μg/kg/min. As expected, based on this unique adrenergic receptor pharmacology, dobutamine enhances myocardial contractility and modestly reduces arterial vasomotor tone through activation of β1 and β2 adrenoceptors, respectively. These properties combine to substantially increase cardiac output in the presence or absence of LV dysfunction. Notably, the negative isomer of dobutamine begins to stimulate the α1 adrenoceptor at infusion rates >5 μg/kg/min, an action that limits the magnitude of β2 adrenoceptor-mediated vasodilation. This effect preserves LV preload, afterload, and arterial pressure; sustains increases in cardiac output; and may serve to attenuate profound baroreceptor reflex–mediated tachycardia that might otherwise occur. Despite this latter effect, dobutamine often markedly increases heart rate by direct chronotropic effects resulting from β1
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


