FIGURE 12-1 A: Representative neuron and myelinated and nonmyelinated axon. The neuron consists of a cell body (soma), dendrites, and an axon. Myelinated nerve fibers have a sheath composed of a continuous series of neurolemma (derived from Schwann cells) that surround the axon and form a series of myelin segments. Multiple nonmyelinated nerve fibers are individually encased within a single neurolemma that does not produce myelin. B: Arrangement of perineural connective tissue layers in a representative nerve. Peripheral nerves consist of bundles of nerve fibers, the layers of the connective tissues (endoneurium, perineurium, and epineurium) that serve to bind them, and associated blood vessels (vasa nervorum) that supply them. All but the smallest peripheral nerves are arranged in bundles called fascicles.
Peripheral nerve fibers are organized within three layers of connective tissue (Fig. 12-1B). Individual nerve fibers are immediately surrounded by endoneurium, consisting of delicate connective tissue that consists of Schwann cells and fibroblasts along with capillaries. A dense layer of collagenous connective tissue, the perineurium, encloses bundles of nerve fibers into a fascicle. It functionally provides an effective barrier against penetration of the nerve fibers by foreign substances. The epineurium is also a dense connective tissue layer that surrounds and encases bundles of fascicles together into a cylindrical sheath structurally similar to a coaxial cable. An additional connective tissue layer that forms a paraneural sheath further encases peripheral nerves. Together, these tissue layers offer protection to peripheral nerves but also present a significant barrier to passive diffusion of local anesthetics toward the axonal cell membrane.
Peripheral nerves are mixed nerves containing both afferent and efferent nerve fibers that are either myelinated or nonmyelinated (Fig. 12-1A). The cell membrane (neurolemma) of Schwann cells envelops axons. Nonmyelinated nerve fibers consist of multiple axons that are simultaneously encased by the neurolemma of a single Schwann cell. Voltage-gated sodium channels (VGNa) are uniformly distributed along the entire axon of nonmyelinated nerve fibers. In contrast, a myelinated nerve fiber is segmentally encased by a myelin sheath that is derived from a continuous series of neurolemma that concentrically wraps around a single axon. Specialized regions, known as the nodes of Ranvier, where the VGNa are concentrated along the axons of myelinated nerve fibers, periodically interrupt the myelin sheath. Along myelinated axons, Na+ conductance is restricted to the nodes of Ranvier. This allows action potential propagation to jump from one node to the next via saltatory conduction, which significantly enhances the speed of signal transmission (Table 12-1).
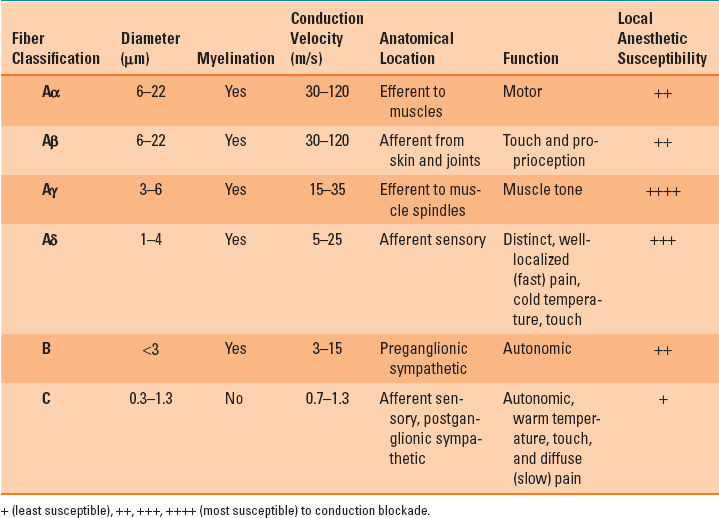
Table 12-1 Classification of Peripheral Nerve Fibers
B. Electrophysiology of Neural Conduction and Voltage-gated Sodium Channels
Neurons maintain a resting membrane potential of approximately –60 to –70 mV. The Na+-K+ (potassium) pump actively cotransports three Na+ ions out of the cell for every two K+ ions into the cell. This creates an electrochemical concentration gradient across the semipermeable cell membrane. The resulting ionic disequilibrium favors the movement of Na+ ions into the cell and K+ ions out of the cell. However, despite the concentration gradient for both ions, the resting cell membrane is relatively more permeable to K+ ions. This facilitates a net passive efflux of K+ ions out of the cell and leaves a relative net excess of negatively charged ions (polarized) within the axoplasm.
Neural impulses are conducted along axons as action potentials, which are transient membrane depolarizations initiated by various mechanical, chemical, or thermal stimuli. Depolarization is mediated primarily via rapid intracellular influx of Na+ ions flowing down its electrochemical gradient through VGNa. The VGNa spans the axonal membrane and consists of an α-subunit and one or two varying auxiliary β-subunits. The α-subunit forms the ion-conducting pore of the VGNa and it comprises four homologous domains (I to IV), each with six α-helical transmembrane segments. The loops that link the S5 and S6 segments of the α-helices of each of the four domains are located extracellular, extending inward to form the narrowest section of the channel pore. They are believed to provide its ion selectivity.
At the resting membrane potential, the channel pore is in a resting (closed) conformation. Upon an initial depolarization, movement of the S1-S4 voltage-sensing segments leads to rearrangement of the S6 segment. This results in activation (opening) of the channel pore, inducing a sudden increase in Na+ ion permeability. The resultant rapid inward Na+ current activates and opens additional VGNa. This further accelerates depolarization until a threshold membrane potential is reached, triggering an action potential. During the depolarization phase, the inward Na+ current flows into the axoplasm and spreads to the adjacent (inactive) cell membrane, resulting in a wave of sequential depolarization (and the action potential) propagating along the axon. Although the wave of depolarization spreads from the initial area of excitation in both directions, the just activated membrane behind the impulse is temporarily refractory to subsequent depolarization. Thus, the propagation of the impulse is unidirectional. The activated VGNa is inactivated within milliseconds by an additional conformational change. This leads to binding of the cytoplasmic loop located between domains III and IV to the cytoplasmic opening of the VGNa to form the rapid inactivation gate. The rapid inactivation gate functions as an intracellular blocking particle that folds into and blocks the channel pore. This rapid inactivation process is required for repetitive firing of action potentials in neural circuits and for control of excitability in neurons. Repolarization occurs due to a combination of a progressive decrease in the driving force for the inward Na+ current and inactivation of VGNa. In addition, membrane depolarization simultaneously activates voltage-gated K+ channels. This leads to an outward positive current of K+ ions, which in conjunction with VGNa inactivation, eventually returns the axonal membrane to or just beyond (hyperpolarization) its resting membrane potential. In summary, inward positive currents, mediated by Na+ ions, depolarize the membrane, and in contrast, outward positive currents, mediated by K+ ions, repolarize the membrane.
C. Voltage-gated Sodium Channels and Interactions with Local Anesthetics
Local anesthetics act at the axonal membrane by binding to a specific region within the α-subunit. This prevents VGNa activation, thus inhibiting the inward Na+ current that mediates membrane depolarization. The binding site for local anesthetics is located within the channel pore and is formed from amino acid residues in the S6 segments of domains I, III, and IV. The binding site may be approached from two pathways: from the intracellular aspect of the channel pore (hydrophilic pathway) or laterally from within the lipid membrane (hydrophobic pathway). As the amount of administered local anesthetic increases, an increasing percentage of VGNa bind to local anesthetics, further inhibiting the inward Na+ current. Subsequently, the rate of depolarization (in response to stimulation) is attenuated, inhibiting the achievement of the threshold membrane potential. Consequently, achievement of an action potential becomes increasingly difficult. With a sufficient number of local anesthetic-bound VGNa, an action potential can no longer be generated and impulse propagation is blocked. Local anesthetic binding to VGNa does not alter the resting membrane potential nor does it alter the threshold potential.
Local anesthetics bind more avidly to VGNa in the activated (open) and inactivated (channel pore is open but closed by movement of the inactivation gate) conformations. The difference in binding affinity is attributable to the difference in the availability of the two pathways for local anesthetic to reach the binding site. Local anesthetics produce a concentration-dependent decrease in inward Na+ current characterized as tonic blockade, representing a decrease in the number of open confirmation VGNa (1). With repeated depolarization, a greater number of VGNa are in either the activated or inactivated conformations. Therefore, they can be bound at a given local anesthetic concentration. Additionally, the dissociation rate of local anesthetics from their binding site is slower than the rate of transition from the inactivated to the resting conformation. Thus, repeated stimulation results in accumulation of local anesthetic-bound VGNa characterized as frequency-dependent blockade.

Not only do local anesthetics prevent nerve impulse propagation by adhering to binding sites on voltage-gated sodium channels in the cell membrane (tonic blockade), but they also dissociate from the binding site more slowly than the site can return to its resting conformation (frequency-dependent blockade).
D. Mechanisms of Nerve Block
In order for local anesthetics to bind VGNa, they must reach the neural membrane. Thus, local anesthetics must penetrate through variable amounts of perineural tissue and still maintain a sufficient concentration gradient to diffuse through the lipid bilayer. Only a small fraction (1% to 2%) of local anesthetic reaches the neural membrane even when deposited in close proximity to peripheral nerves. Peripheral nerves that have been desheathed in vitro require about a hundredfold lower local anesthetic concentration than peripheral nerves in vivo. In contrast, central neuraxial nerves are encased in three layers of meninges: the pia mater, arachnoid membrane, and dura mater. The pia mater is adherent to the nerves themselves and is separated from the arachnoid membrane by cerebrospinal fluid that fills the space between these two layers. The subarachnoid space, where the spinal nerves are only covered by the pia mater, is the target location for spinal anesthesia. The dura mater further encases the arachnoid membrane, forming the dural sac, a tough covering around the central neuraxis. The epidural space consists of everything located within the vertebral canal but outside the dural sac. The presence of the arachnoid membrane and dura mater result in tenfold higher local anesthetic dose requirements to produce complete epidural blockade compared with that required in the subarachnoid space.
The quality of nerve block is determined not only by the intrinsic potency of the chosen local anesthetic but also by the concentration and volume of the administered local anesthetic. The potency of a local anesthetic can be expressed as the minimum effective concentration required to establish complete nerve blockade. The volume of local anesthetic is also important, as a sufficient length of axon or successive nodes of Ranvier must be blocked in order to inhibit regeneration of the neural impulse. This is due to the phenomenon of decremental conduction. Membrane depolarization passively decays with distance away from the front of the action potential to the point that impulse propagation stops when depolarization falls below the threshold for VGNa activation. If less than a critical length of axon is blocked, the action potential may still be regenerated in the proximal neural membrane segment or node of Ranvier when the decaying depolarization is still above the threshold potential.
Different types of nerve fibers demonstrate varying minimal blocking concentrations and local anesthetic susceptibilities (Table 12-1). Clinically, there is a predictable progression of sensory and motor function blockade, starting first with loss of temperature sensation, followed by proprioception, motor function, sharp pain, and lastly light touch. Termed differential block, this progression was initially attributed to differences in axon diameter, with smaller fibers inherently more susceptible to conduction blockade compared with that of larger fibers. However, small myelinated fibers (Aγ and Aδ) are the most susceptible to conduction blockade. Next in order of block susceptibility are large myelinated fibers (Aα and Aβ), and the least susceptible are small, nonmyelinated C fibers.

Differential blockade refers to the predictable progression of sensory and motor function blockade by local anesthetics, starting with loss of temperature sensation and followed by impaired proprioception, motor function, sharp pain, and lastly light touch.
Within peripheral nerves, longitudinal and radial diffusion of local anesthetic will produce varying drug concentrations along and within the nerve during the onset and recovery from clinical block. When local anesthetics are deposited around a peripheral nerve, diffusion progresses from the outer surface (mantle) toward the center (core) along a concentration gradient. Consequently, nerve fibers arranged in the mantle of mixed peripheral nerves are blocked initially. These outer nerve fibers are typically distributed to more proximal anatomic structures, whereas core fibers innervate more distal structures. This topographical arrangement explains the initial development of proximal, followed by distal anesthesia, as local anesthetic diffuses to the more centrally located core nerve fibers. In summary, the sequence of onset and recovery from peripheral nerve block depends on a combination of the topographical arrangement of the nerve fibers within a mixed peripheral nerve and their inherent susceptibility to local anesthetic blockade.
II. Local Anesthetic Pharmacodynamics
A. Physiochemical Properties and Relationship to Activity and Potency
Local anesthetics in solution are weak bases that typically carry a positive charge at the amine group at physiologic pH. The prototypical local anesthetic structure consists of a hydrophobic group (typically a lipid-soluble aromatic ring) connected to a hydrophilic group (charged amine) by either an amide or ester linkage (Fig. 12-2). The nature of the chemical bond is the basis for classification of local anesthetics as either an aminoamide or aminoester (Table 12-2). Although the nature of the linkage determines the basis for metabolism (aminoamides are metabolized in the liver, whereas aminoesters are metabolized by plasma cholinesterase), the physiochemical properties are largely determined by the nature of the alkyl substitutions on either the aromatic ring or the amine group, the charge of the amine group, or the stereochemistry of the related isomers (Table 12-2). These physiochemical properties largely determine the potency, onset and duration of action, and tendency for differential nerve block.

Although the linkage between a local anesthetic’s hydrophobic aromatic ring and its hydrophilic amine group determines the molecule’s class (aminoamide or aminoester) and metabolic pathway, it is the chemical substitutions on the aromatic ring or amine group that determine the drug’s potency, onset, and duration of action.
Table 12-2 Chemical Structure and Physiochemical Properties of Clinically Useful Local Anesthetic Agents
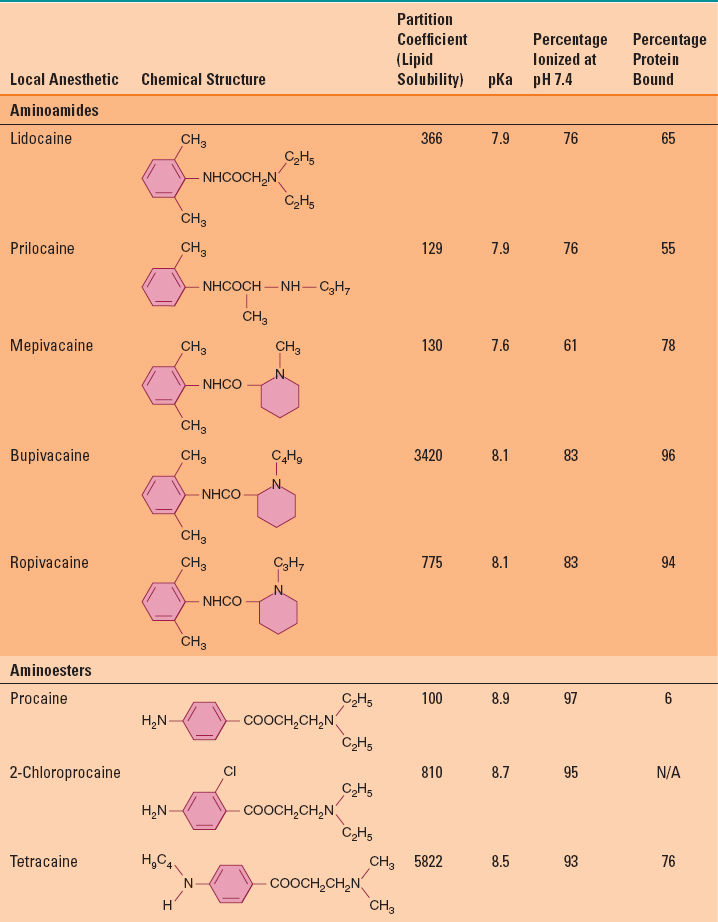
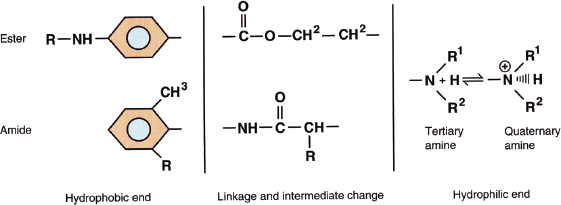
FIGURE 12-2 The prototypical structures of aminoester and aminoamide local anesthetics. (From Mulroy F, Bernards CM, McDonald SB, Salinas FV. A Practical Approach to Regional Anesthesia. 4th Edition. Philadelphia: Wolters Kluwer, 2009:2.)
Lipid solubility is determined by the degree of alkyl substitutions on either the aromatic ring or the amine group. Lipid solubility is typically expressed by the partition coefficient in a hydrophobic solvent (typically octanol). Compounds with increased octanol solubility are more lipid soluble (Table 12-2). Increased lipid solubility enhances the ability to penetrate the lipid membrane and deliver local anesthetic in closer proximity to the membrane bound VGNa, which in turn correlates with the potency and, to a lesser extent, the duration of action. Although lipid solubility correlates with octanol solubility (and inherent potency in vitro), the minimum in vivo local anesthetic concentration that will block impulse conduction may be affected by numerous factors such as fiber size, type, and myelination, tissue pH (see below), local tissue redistribution and sequestration into lipid-rich perineural compartments, and inherent vasoactive properties of the specific local anesthetic.
At physiological pH, local anesthetics are weak bases that exist in equilibrium between either the lipid soluble base form or the water-soluble ionized form. The relative percentage of each form is determined by the dissociation constant (pKa) and surrounding tissue pH. The pKa is the pH at which the percentage of each form is equal (Table 12-2), which is defined by the Henderson-Hasselbalch equation:
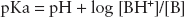
where [BH+] is the concentration of the charged, lipid-insoluble form of the local anesthetic, and [B] is the concentration of the uncharged lipid-soluble form of local anesthetic.
The lower the pKa for a given local anesthetic, the higher the percentage of the lipid-soluble base form that exists to more readily penetrate the lipid cell membrane, thus speeding the onset of action. After penetration through the cell membrane into the axoplasm, equilibrium between the base form and the charged form is re-established. It is the charged form within the axoplasm that more avidly binds to local anesthetic binding sites within the channel pore of the VGNa.
The majority of clinically useful local anesthetics are formulated as racemic compounds. These are one-to-one mixtures of enantiomeric stereoisomers bearing identical chemical composition, but with a different three-dimensional spatial orientation around an asymmetric carbon atom. Although enantiomers of local anesthetics have identical physiochemical properties, they exhibit different clinical pharmacodynamics (potency) due to subtle differences in interaction and binding of VGNa
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


