Babak Sadighi
Jessica Black
Before examining any anesthetic medication in detail, it is important to consider what occurs when an intravenous sedative is administered. Unlike inhaled agents, the mechanisms of action for intravenous sedatives and anesthetics are well characterized.
Initially, the medication will be transported and diluted within the patient’s cardiovascular system. There will be no change in the patient’s level of consciousness. Some of the medication will be transported across the blood–brain barrier where it will become redistributed to its effect site and bind to the receptors for which it has affinity.
As the medication binds to its target receptors, the onset of sedation can be observed. There will be a progressive diminution in the patient’s level of consciousness, continuing until chemical equilibrium is reached at the effect site. Depending on the medication and the quantity administered, this decreased level of consciousness may range from light sedation with preserved spontaneous ventilation to general anesthesia with apnea.
The medication that is circulating in the cardiovascular system is eventually eliminated from the body, metabolized into an inactive or degraded form, or redistributed and sequestered into other peripheral tissues in which it produces no effect. The concentration of the medication within the cerebral circulation will start to fall, and the concentration gradient will draw the medication away from its effect site. Binding at the target receptors within the brain then decreases, and recovery toward consciousness begins.
If it is desired to prolong the patient’s sedation, additional medication can be administered. In response to these further doses, the concentration of medication will instead approach a new equilibrium concentration with a corresponding level of clinical sedation. Eventually, the patient can be allowed to recover by discontinuing the infusion. The additional medication administered must then be eliminated, metabolized, or redistributed. The medication will return into circulation from the peripheral tissues into which it had been sequestered. The quantity of medication present may also temporarily exceed the metabolic capacity of the pathways by which it is degraded. Consequently, the clinical return to consciousness will now be slower than before and will depend on the quantity and duration of the medication’s administration. Finally, it must be considered that an individual may not respond as expected or may have an unanticipated adverse reaction to the medication instead.
In this vignette, there are four pharmacologic themes, illustrating the clinical use of an intravenous anesthetic or sedative:
1. Mechanism of action: molecular interaction of a medication with a receptor
2. Pharmacokinetic profile: the trafficking, redistribution, and elimination of the medication within the body
3. Pharmacodynamic profile: the behavior of a concentration of this medication at the population of receptors within the brain
4. Adverse reactions: the extent to which the medication may produce allergic or hypersensitivity reactions.
I. General Pharmacology of Intravenous Anesthetics
A. Mechanism of Action
The most commonly used intravenous anesthetic agents—the barbiturates, propofol, the benzodiazepines, and etomidate—all act at the site of the γ-aminobutyric acid A (GABAA) receptor, as shown schematically in Figure 9-1. GABA is the main inhibitory neurotransmitter within the central nervous system, and its action at the GABAA receptor causes increased transport of chloride (Cl−) ions across the membrane and into the postsynaptic neuron. The postsynaptic neuron becomes hyperpolarized, which functionally inhibits further propagation of nerve signals. The GABAA receptor is therefore a ligand-activated ion channel composed of five subunits. Intravenous anesthetics that bind to the GABAA receptor do not bind at the same location as GABA itself (the orthosteric binding site), instead they bind at other locations (allosteric sites) and change the effect of GABA upon the receptor. These intravenous anesthetics are therefore positive allosteric modulators of the GABAA receptor and cause receptor conformational changes such that the action of GABA itself is potentiated and sedation occurs. The subunit composition of GABAA receptors can vary: there are 19 different possible subunits arising from eight different subunit classes (α1–6, β1–3, γ1–3, δ, ε, θ, π, and ρ). Intravenous anesthetic agents may only be active at receptors expressing certain combinations: the benzodiazepine allosteric binding site occurs only at the interface of α and γ2 subunits, and etomidate is active primarily at GABAA receptors that contain β2 or β3 subunits.

GABA is the main inhibitory neurotransmitter within the central nervous system. Its action at the GABAA receptor causes increased transport of chloride (Cl−) ions across the membrane, causing it to become hyperpolarized.
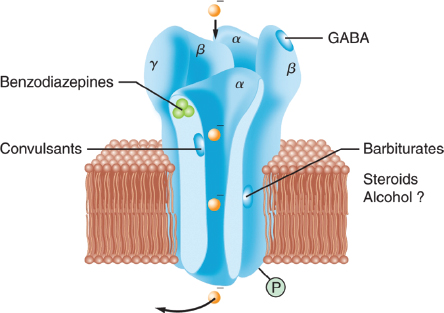
Figure 9-1 Schematic model of the γ-aminobutyric acid A (GABAA) receptor complex, illustrating recognition sites for many of the substances that bind to the receptor. (From White PF, Eng MR. Intravenous anesthetics. In: Barash PG, Cullen BF, Stoelting RK, et al. Clinical Anesthesia. 7th ed. Philadelphia: Wolters Kluwer Health/LWW, 2013:480, with permission.)
 VIDEO 9-1
VIDEO 9-1
GABA-A Receptors
B. Pharmacokinetics and Metabolism
The redistribution and elimination of intravenous anesthetics within the body can be approximated with a simplified three-compartment model of the body. In this model, medications are administered into a first well-mixed central compartment. Diffusion occurs back and forth between this first compartment and the additional second and third peripheral compartments. The diffusion constants (shown as k in Fig. 9-2) are such that one peripheral compartment equilibrates quickly with the central compartment and one equilibrates more slowly. The drug is not pharmacologically active in these peripheral compartments. Instead, they act as reservoirs into which medications are redistributed and sequestered. These peripheral compartments model how the action of the medication may be terminated by redistribution. They also model the way in which accumulation of medication within these peripheral compartments can lead to progressively increasing context-sensitive half-times as medication diffuses back into the central compartment. An effect site compartment models the receptor population at which the medication has its mechanism of action. Diffusion also occurs between the central and the effect site compartments. But because the quantity of drug bound to the receptors at any given moment is minor compared with the total quantity of medication in the body, the effect site compartment is assumed to be sufficiently small that its effect on the mass of medication within the central compartment is negligible.
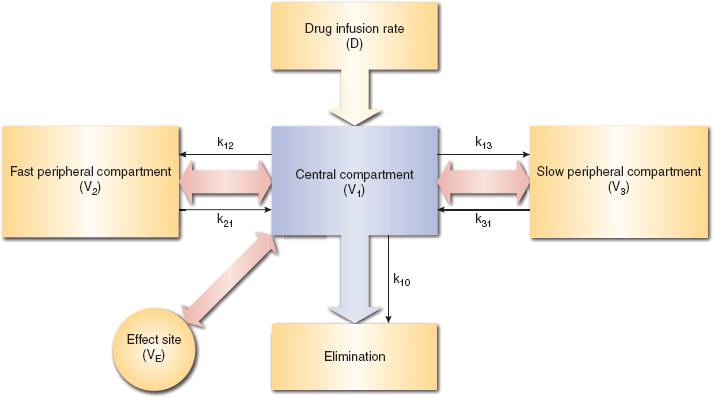
Figure 9-2 Three-compartment model for the pharmacokinetic modeling of intravenous medication administration, redistribution, and elimination. Additionally, an effect site compartment is present. The volume of this compartment is assumed to be sufficiently small that the effect on the quantity of medication in the central compartment is negligible.
The structure of these compartments, with their associated volumes and diffusion coefficients, allows a series of differential equations to be produced that model the trafficking of medication in response to changes in infusion rates. A computer can be used to determine an ideal sequence of infusion rate changes so that the concentration of drug at the effect site is brought to the desired concentration within the optimum time (1). This practice is known as target-controlled infusion.
C. Pharmacodynamic Effects
Many combinations of GABAA receptor subunits are possible. However, in practice, these variations are not directly accounted for between individual receptors when predicting the clinical effect of a dosage of a medication. Instead, the effect site in the pharmacokinetic model represents the combined population of all the receptors, and the likely clinical effect is determined by a statistical model relating this concentration to a particular clinical outcome. These pharmacodynamic models are created for different clinical outcomes with different medications. Figure 9-3 shows two separate pharmacodynamic models: the leftmost model relates the concentration of propofol at the effect site to the loss of the eyelash reflex, and the rightmost model relates the concentration of propofol to loss of consciousness (2). The most notable characteristic is the strong nonlinearity of these relationships. It is possible, with only a small increase in concentration of propofol, to transition rapidly from consciousness to unconsciousness. These nonlinear models have a standard form, known as the sigmoid-Emax model or the Hill equation (3):
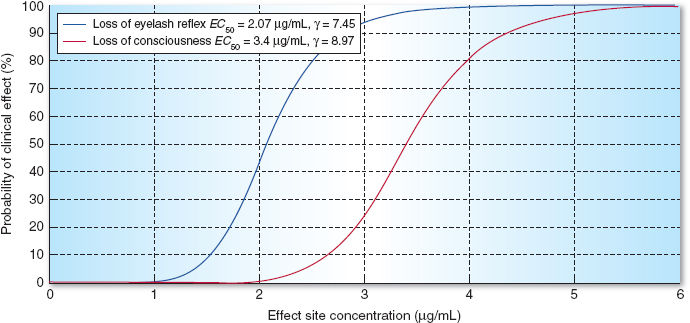
Figure 9-3 Pharmacodynamic models for the probability of loss of eyelash reflex and for the probability of loss of consciousness based on propofol effect site concentration.
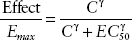
The variable EC50 is the concentration at the effect site at which 50% of the maximal effect is seen. This is a measure of the potency of the medication. The γ variable is the sigmoid coefficient, and greater values of γ yield more abrupt transitions. A small change in propofol effect site concentration can cause a large, even unexpected, clinical effect.
D. Hypersensitivity (Allergic) Reactions
The most common causes of hypotension following induction with intravenous anesthetic agents are unrecognized hypovolemia and unexpected drug interactions. True hypersensitivity reactions are rare, although case reports of histamine release with all intravenous anesthetic agents, with the exception of etomidate, have been alleged. Propofol does not normally cause histamine release but anaphylactoid reactions have been reported in patients with multiple drug allergies. Barbiturates can precipitate acute intermittent porphyria in susceptible patients.
II. Comparative Physiochemical and Clinical Pharmacologic Properties
A. Barbiturates
The most commonly used barbiturates are the thiobarbiturates: thiopental (Pentothal, 5-ethyl-5-[1-methylbutyl]-2-thiobarbituric acid), thiamylal (Surital, 5-allyl-5-[1-methylbutyl]-2-thiobarbituric acid), and the oxybarbiturate methohexital (Brevital, 1-methyl-5-allyl-5-[1-methyl-2-pentanyl] barbituric acid).
Barbiturates depress the reticular activating system in the brainstem and are believed to potentiate the action of GABAA receptors, increasing the duration of an associated chloride ion channel opening. Barbiturates decrease cerebral metabolic rate of oxygen (CMRO2), cerebral blood flow (CBF), and intracranial pressure (ICP). Barbiturates can induce an isoelectric electroencephalogram (EEG), maximally decreasing CMRO2.

Commonly used intravenous anesthetic agents such as the barbiturates, propofol, the benzodiazepines, and etomidate act at the site of the GABAA receptor.
Barbiturates are formulated as sodium salts and are reconstituted in water or isotonic sodium chloride (0.9%) to prepare 2.5% thiopental, 1% to 2% methohexital, and 2% thiamylal. These preparations are highly alkaline (pH 9 to 10). When they are added to lactated ringers or other acidic drug preparations, crystalline precipitation will occur and may irreversibly occlude intravenous tubing and catheters. Barbiturates rarely cause pain on injection but will cause significant tissue irritation if extravasated. Inadvertent intra-arterial injection of thiobarbiturates causes serious complications, including intense vasoconstriction, thrombosis, and tissue necrosis. Immediate treatment may require intra-arterial papaverine and lidocaine or procaine, regional anesthesia-induced sympathectomy (stellate ganglion block, brachial plexus block), and heparinization.
The anesthetic action of the barbiturates is primarily terminated by redistribution from the central lipophilic tissues of the brain to peripheral lean muscle compartments. Barbiturates undergo slow terminal elimination via hepatic metabolism, biliary conjugation, and renal excretion. The terminal elimination of thiopental is prolonged with a half-life of 10 to 12 hours. Methohexital clearance is more dependent on hepatic blood flow, allowing for a shorter elimination half-life of 4 hours.
Care should be taken in patients with porphyrias because barbiturates stimulate porphyrin formation and can precipitate an acute crisis.
B. Propofol
Propofol (Diprivan) is an alkylphenol compound prepared in an egg lecithin emulsion consisting of soybean oil, glycerol, egg phosphatide, and ethylenediaminetetraacetic acid or metabisulphite as an antimicrobial.
 VIDEO 9-2
VIDEO 9-2
Propofol
Propofol increases the binding affinity of GABA with the GABAA receptor. Coupled to a chloride channel, the activation leads to the hyperpolarization
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


