Diseases of the Adrenal Glands
Muriel N. Nathan PhD, MD
The adrenal glands, which sit on top of the kidneys, are very small organs with complex functions. The gland has a cortex that secretes glucocorticoids and mineralocorticoids. The medulla of the adrenal gland produces catecholamines, epinephrine, and norepinephrine. Each of these will be discussed in detail. Diseases of the adrenal glands can lead to syndromes of wasting, hypotension, and hypoglycemia if adrenal hormones are lacking, whereas diseases that increase the production of adrenal hormones can produce hypertension, weight gain, and virilization. The following chapter will give the primary care provider information needed for the diagnosis and management of several adrenal diseases: adrenal insufficiency, adrenal overactivity, hyperaldosteronism, hypoaldosteronism, and pheochromocytoma. Appropriate referral information will also be given.
ANATOMY, PHYSIOLOGY, AND PATHOLOGY
The adrenal glands are small pyramidal organs, measuring 3 × 6 cm and weighing about 2.5 to 5 g each, embedded in the retroperitoneal fat medial to the superior pole of the kidneys (Rittmaster & Arab, 1995). The blood supply branches off multiple sites; vascular sites drain from the suprarenal vein into the inferior vena cava on the right and the renal vein on the left. Autonomic innervation of the glands is extensive.
Histologically, the cortex is composed of three parts: the zona glomerulosa, which secretes mineralocorticoids, and the zona fasciculata and zona reticularis, which secrete glucocorticoids and androgens. The medulla arises from neural crest cells. It is very vascular and consists of chromaffin cells, arranged in a network, that secrete catecholamines (Rittmaster & Arab, 1995).
When adrenal loss is caused by autoimmune disease, the cortices of the adrenal glands are decreased in size and infiltrated by lymphocytes. Normal adrenal architecture is replaced by fibrosis. When adrenal failure is caused by tuberculosis or fungal infection, the glands are often large and calcified. Adrenal insufficiency can also occur secondary to hemorrhage within the gland, which has a plentiful arterial supply but limited venous drainage, increasing the risk of thrombosis (Rittmaster & Arab, 1995; Loriaux, 1995).
Synthesis of the steroid hormones is regulated by the hypothalamic–pituitary–adrenal axis (HPA) and, with the exception of aldosterone from the zona glomerulosa, is dependent on adrenocorticotrophic hormone (ACTH). ACTH is produced in the anterior pituitary. Its regulation is maintained by the feedback inhibition of cortisol. In addition, corticotropin-releasing hormone (CRH) stimulates ACTH release. Circadian rhythms and stress also influence the release of ACTH. Cortisol, when present in higher-than-physiologic levels, will inhibit ACTH secretion (negative feedback loop). ACTH and cortisol have a diurnal variation, with maximal secretion between 2 AM and 8 AM and nadir around midnight. ACTH secretion is also increased by fever, hypoglycemia, pregnancy, strenuous exercise, anorexia nervosa, and depression (White et al, 1995).
Corticotropin (ACTH) can stimulate aldosterone secretion, but its effect is short-lived. Aldosterone is instead regulated by the renin–angiotensin system and potassium. Receptors in the wall of the afferent arteriole of the kidney respond to hypovolemia and stimulate the release of renin from the juxtaglomerular cells. This cleaves angiotensinogen, a protein made by the liver and found in the plasma, to angiotensin I. Angiotensin-converting enzyme (ACE), elaborated by pulmonary endothelial cells, then cleaves angiotensin I, a decapeptide, to angiotensin II, an octapeptide. Angiotensin II binds to a membrane receptor on the zona glomerulosa cells that is in the family of G-protein-type receptors. Activated second messengers raise intracellular calcium concentrations, leading to aldosterone secretion. Potassium can also increase aldosterone secretion via depolarization of cell membranes, leading to calcium influx through voltage-gated channels (White, 1994).
The primary precursor of adrenal steroid hormones is cholesterol, which is stored in the adrenal cortex. Multiple enzymatic steps produce the steroid hormones along three pathways, producing mineralocorticoids, glucocorticoids, and androgens.
ADRENAL INSUFFICIENCY
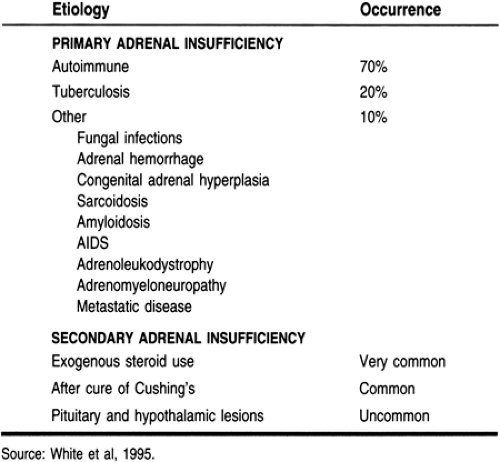
Adrenal insufficiency results from a decreased concentration of glucocorticoids, such as cortisol. The most common cause is the withdrawal of exogenous steroids, often given to treat asthma or rheumatologic conditions. Primary adrenal insufficiency is caused by loss of the adrenal cortex; more than 90% of the cortex must be destroyed for adrenal insufficiency to occur (Loriaux, 1995). Secondary adrenal insufficiency is the loss of cortisol secretion because of loss of pituitary ACTH secretion (Oelkers, 1996). Causes of adrenal insufficiency are presented in Table 18-1.
 |
Epidemiology
Primary adrenal insufficiency is caused by autoimmune destruction of the adrenal cortex in industrialized countries. About 70% of the cases of adrenal loss (40 to 60 per 1 million in North America) involve autoimmune adrenal insufficiency or Addison’s disease. This can occur from age 17 to 72, but usually presents by age 40. It is three times more common in women
than men. Half of the patients with autoimmune adrenal insufficiency have other autoimmune illnesses, such as early gonadal failure, insulin-dependent diabetes mellitus, Hashimoto’s hypothyroidism or Graves’ hyperthyroidism, hypoparathyroidism, or pernicious anemia. Autoimmune adrenal insufficiency is called Addison’s disease. Schmidt’s syndrome is the combination of autoimmune adrenal insufficiency and diabetes with or without hypothyroidism. When more than two autoimmune diseases coexist, the syndrome is called polyglandular autoimmune syndrome, where antibodies to 21-hydroxylase (an enzyme involved in steroid synthesis) are found in the blood (Loriaux, 1995; Oelkers, 1996; Rao et al, 1989).
than men. Half of the patients with autoimmune adrenal insufficiency have other autoimmune illnesses, such as early gonadal failure, insulin-dependent diabetes mellitus, Hashimoto’s hypothyroidism or Graves’ hyperthyroidism, hypoparathyroidism, or pernicious anemia. Autoimmune adrenal insufficiency is called Addison’s disease. Schmidt’s syndrome is the combination of autoimmune adrenal insufficiency and diabetes with or without hypothyroidism. When more than two autoimmune diseases coexist, the syndrome is called polyglandular autoimmune syndrome, where antibodies to 21-hydroxylase (an enzyme involved in steroid synthesis) are found in the blood (Loriaux, 1995; Oelkers, 1996; Rao et al, 1989).
The second leading cause of adrenal insufficiency, found in 20% of those with adrenal loss, is cortical destruction from infectious diseases, such as tuberculosis or fungal infections. In developing countries, tuberculosis rather than autoimmune disease is the leading cause of adrenal insufficiency. Adrenal failure can be seen in up to 5% of patients with AIDS, usually late in the course of the disease and a result of cytomegalovirus infection (Loriaux, 1995).
Other causes of adrenal failure include hemorrhage in the adrenal gland (Loriaux, 1995; Oelkers, 1996; Rao et al, 1989). Unrecognized, this can lead to shock and death, which is then incorrectly attributed to sepsis or ischemia. This is seen in patients who have hypercoagulable states such as lupus, diabetes, or pregnancy, patients using anticoagulants, or patients in the postoperative period. This can also be seen during sepsis, particularly meningococcemia, pneumococcal pneumonia, or Haemophilus influenzae (Friedrich-Waterhouse syndrome). Patients with antiphospholipid syndrome, often associated with lupus, can also lose adrenal function because of arterial and venous thrombi (Oelkers, 1996).
Drugs that work by interfering with steroid synthesis or that accelerate steroid degradation can cause loss of adrenal function. These drugs include those used to treat excessive steroid production (ie, Cushing’s syndrome) and adrenal carcinoma, such as aminoglutethimide, mitotane (o,p′-DDD, related to the insecticide DDT), and ketoconazole. The latter, for example, inhibits two enzymes in the glucocorticoid-synthetic pathway and binds to the glucocorticoid receptor. Drugs such as rifampin, phenytoin, and phenobarbital accelerate the catabolism of cortisone by stimulating hepatic microsomal enzymes. Their use causes adrenal insufficiency in patients with compromised adrenal function (Loriaux, 1995).
Rarer causes of adrenal cortical loss include metastatic disease, although adrenal failure is rare. This can occur in up to 58% of women with breast cancer, 42% of those with lung cancer, and 50% of those with malignant melanoma. Other infiltrating diseases, such as amyloidosis, sarcoidosis, or hemochromatosis, can lead to loss of adrenal cortical function. Familial disorders—X-linked or autosomal recessive metabolic disorders, adrenoleukodystrophy, and the milder adrenomyeloneuropathy—cause adrenal deficiency primarily in young men (Loriaux, 1995).
History and Physical Examination
Adrenal insufficiency can present as an insidious process or as an acute crisis (Loriaux, 1995; Oelkers, 1996). Patients complain of anorexia, weight loss, and weakness (Table 18-2). Fatigue, sweating, and loss of concentration can occur, especially as the loss of glucocorticoids enhances hypoglycemia. About half the patients have gastrointestinal complaints, usually nausea, vomiting, abdominal or loin pain, and diarrhea. In primary adrenal insufficiency, the patient notes darkening of the skin, caused by enhanced secretion of pro-opiomelanocortin-derived peptides. The increased pigmentation is found at the elbows, knees, creases of the palmar surface, gingival margin, or buccal mucosa. Vitiligo is also seen. Hypotension, with systolic readings under 110 mmHg, and orthostatic blood pressure changes are found. Women report hair thinning and irregular menses. Psychiatric symptoms are seen in 64% to 84% of patients with adrenal insufficiency. The most common findings are depression, apathy, or confusion, but there are reports of psychosis, paranoia, schizophrenia, and self-mutilation (Loriaux, 1995).
 |
Adrenal hemorrhage with loss of adrenal function can be a complication of several illnesses, including sepsis, trauma with shock, coagulopathies, and ischemic disorders. The clinical signs can be vague, but usually patients have abdominal, flank, back, or chest pain with fever and hypotension. Anorexia, vomiting, psychiatric symptoms, and abdominal rigidity with rebound also occur (Loriaux, 1995; Rao et al, 1989).
Pituitary or hypothalamic disease can lead to secondary adrenal failure. Secondary adrenal failure is often associated with other secondary losses, such as hypothyroidism or hypogonadism (Loriaux, 1995; Oelkers, 1996). Thus, patients may present with fatigue, loss of menses, loss of libido, or difficulty getting
erections. In secondary adrenal insufficiency, aldosterone secretion is preserved because aldosterone is regulated by the renin–angiotensin axis rather than ACTH (White, 1994).
erections. In secondary adrenal insufficiency, aldosterone secretion is preserved because aldosterone is regulated by the renin–angiotensin axis rather than ACTH (White, 1994).
Loss of aldosterone secretion can occur in isolation (hyperreninemic hypoaldosteronism) or as a result of diminished secretion of renin or angiotensin II (hyporeninemic hypoaldosteronism). Hyporeninemic hypoaldosteronism is commonly found in patients with mild renal insufficiency (eg, diabetes [50% of the patients]) or those with nephritis, but it can also be caused by use of nonsteroidal anti-inflammatory drugs (NSAIDs; see section on hypoaldosteronism). Hyperreninemic hypoaldosteronism is usually seen in critically ill patients and may be caused by hypotensive injury to the adrenal glands. It can also be seen in patients with diabetes or those taking heparin (Loriaux, 1995). These patients are usually asymptomatic but can have cardiac arrhythmias or muscle weakness because of hyperkalemia.
Diagnostic Studies
LABORATORY FINDINGS
Sixty-five percent of patients with primary adrenal insufficiency are hyperkalemic, and 90% are hyponatremic from renal salt wasting. In primary but not secondary adrenal insufficiency, mineralocorticoids (aldosterone) are lost along with the glucocorticoid secretion, causing intravascular volume depletion (hypotension) and elevated serum potassium levels. Primary adrenal insufficiency frequently causes hypoglycemia, hypercalcemia, and a mild normocytic anemia with lymphocytosis and mild eosinophilia (Loriaux, 1995; Oelkers, 1996).
In secondary adrenal insufficiency, hyponatremia but not hyperkalemia can occur, but from a different cause. The lack of ACTH leads to low cortisol levels, which increase vasopressin secretion and water retention (Loriaux, 1995; Oelkers, 1996). These patients also show evidence of other pituitary hormone abnormalities, such as elevated prolactin if there is a pituitary tumor, or low levels of thyroid-stimulating hormone, follicle-stimulating hormone, luteinizing hormone, and free thyroxine.
In adrenal hemorrhage, there is a precipitous drop in the hematocrit while the number of white blood cells increases (leukocytosis) (Loriaux, 1995). Hyponatremia and hyperkalemia with renal insufficiency (azotemia) and acidosis are also seen.
The definitive diagnosis of adrenal insufficiency is the lack of response of the adrenal gland to ACTH stimulation. Random cortisol levels may not distinguish patients with mild disease from the normal population, but morning cortisol levels greater than 25 mcg/dL (>525 nmol/L) are likely to indicate normal function, whereas patients with 8 to 9 AM cortisol levels under 4 mcg/dL (83 nmol/L) probably have adrenal insufficiency (Oelkers, 1996). The ACTH stimulation test can be done at any time and consists of giving 250 mcg of synthetic ACTH intravenously or intramuscularly (Oelkers, 1996). To differentiate primary from secondary disease, an ACTH level can be drawn before the synthetic ACTH is given: it will be high in primary disease (> 100 pg/mL or 22 pmol/L) and low in secondary disease (Oelkers, 1996). Alternatively, serum aldosterone could be measured; if low, it is indicative of primary disease, whereas its level is normal in secondary disease.
IMAGING STUDIES
In patients with primary adrenal insufficiency from autoimmune disease, imaging of the adrenal glands is not necessary. In other cases, computed tomography (CT) of the adrenal glands should be done to diagnose fungal disease or cancer (Loriaux, 1995; Oelkers, 1996). In patients with secondary disease from a pituitary or hypothalamic process, magnetic resonance imaging (MRI) of the pituitary and hypothalamic region is done.
Treatment Options, Expected Outcomes, and Comprehensive Management
The usual replacement is cortisone, 12 to 15 mg/m2/day, usually given as 25 mg in the morning and 12.5 mg in the evening to try to mimic the natural circadian rhythm of higher steroid production in the morning. British researchers have shown that there is no advantage to giving the greater dose of steroid in the morning, preferring to give 10 mg three times a day (Loriaux, 1995). If other steroids are used, dosages are adjusted according to the relative potency of the drug. For example, prednisone is about four times as potent as hydrocortisone, so replacement is given as 5 mg in the morning and 2.5 mg in the evening. Because steroids can cause gastritis, weight gain, and osteoporosis, the goal is to give the lowest dosage that relieves symptoms; this can be as low as 15 to 20 mg/day of hydrocortisone (Oelkers, 1996). The steroid replacement dose may be adjusted based on 24-hour measurements of urinary cortisol. Mineralocorticoids are necessary in 75% of patients with primary adrenal insufficiency; the dosage is usually 50 to 200 mcg 9-alpha-fludrocortisol (Florinef) given each morning with a liberal-salt diet. This aldosterone substitute can be adjusted by measuring blood pressure, potassium, and plasma renin activity (PRA), which should be in the upper end of the normal range (Oelkers, 1996).
Teaching and Self-Care
Concern about causing iatrogenic alterations in the pituitary–adrenal axis is widespread because of the common use of glucocorticoids to treat chronic diseases such as arthritis, inflammatory bowel disease, asthma, and emphysema.
CLINICAL WARNING
When the adrenal axis is suppressed from glucocorticoid use, abruptly decreasing or stopping the medication or missing a dose could precipitate an adrenal crisis (adrenal insufficiency) with symptoms of weakness, hypotension, and hypoglycemia.
The degree of suppression of the adrenal axis depends on many factors, such as the type of steroid used, the dosage, and the duration of treatment. Nonetheless, many studies show that suppression is not predictable in individual patients, and thus random cortisol levels are not helpful. Patients taking 20 mg or more of prednisone (or its equivalent) for more than 5 days should be suspected of having adrenal suppression, and thus are at risk for adrenal crisis if the medication is not appropriately weaned. Some patients have symptoms of adrenal insufficiency with normal cortisol and ACTH levels, probably from a sudden
elevation in prostaglandin levels after steroid disuse (steroid withdrawal syndrome) (Schlaghecke et al, 1992).
elevation in prostaglandin levels after steroid disuse (steroid withdrawal syndrome) (Schlaghecke et al, 1992).
Alternate-day therapy is an approach used by many providers to minimize adrenal axis suppression and decrease side effects from glucocorticoids while sustaining therapeutic effects (Schlaghecke et al, 1992). This use of a short-acting steroid (prednisone or methylprednisolone) every 48 hours should be the regimen of choice in patients facing more than a few weeks of glucocorticoid therapy for conditions other than adrenal insufficiency. Those lacking cortisol or ACTH secretion must take replacement daily.
During periods of stress such as febrile illness (temperature >100.4°F), gastroenteritis, or outpatient surgery (eg, tooth extraction), additional steroid coverage is given. The oral dose of steroid can be doubled for the first 2 or 3 days until the underlying fever or “24-hour bug” resolves. If patients are unable to take medications orally, they can be instructed to give glucocorticoid by subcutaneous injection or to use glucocorticoid suppositories for replacement (Oelkers, 1996). Patients may also make changes in their diet and lifestyle to manage symptoms of adrenal insufficiency. These include eating a healthy diet that contains ample fresh fruits and vegetables and is low in simple carbohydrates. Moderate exercise should be encouraged, both for overall good health as well as to aid in stress management. If the latter is a problem, patients may benefit from stress-management techniques that are individualized for them. Patients on steroid replacement should be encouraged to wear a Med-Alert bracelet or to carry a medical identification care stating that they use this medication.
Because steroid-related gastritis can be a problem, patients should be encouraged to avoid smoking and the use of aspirin and ibuprofen and to limit caffeine and alcohol intake. They should be advised to take their steroid with food. Some patients may find that eating small, frequent meals helps their tolerance of steroid medication.
Self-care for bone loss and osteoporosis is covered in Chapter 48.
Referral Points and Clinical Warnings
Chronic adrenal failure may persist for months or years before diagnosis, but acute adrenal insufficiency (adrenal crisis) must be promptly recognized and treated to avoid death, which is usually caused by the underlying illnesses (Loriaux, 1995; Oelkers, 1996; Rao et al, 1989). It can be difficult to diagnose acute adrenal crisis; it is often misdiagnosed as sepsis or an acute abdomen. Acute loss of cortical secretion leads to tachycardia, hypotension and vascular collapse, severe abdominal pain, nausea and vomiting, hyponatremia, and hyperkalemia.
CLINICAL PEARL
The provider should consider referring a patient with suspected adrenal insufficiency to an endocrine specialty practice for diagnosis and management planning.
CLINICAL WARNING
Acutely ill patients may be severely hypotensive. They may need emergency referral.
When a patient with known adrenal insufficiency must undergo emergency surgery, steroid coverage is increased and is given intravenously until oral medication can be reliably taken.
If patients have a history of steroid use, and the length of steroid treatment, the time since last use, or the dosage that was given is not clearly known, the safest and most practical clinical solution is to assume they may be adrenal-insufficient.
CLINICAL WARNING
It is recommended practice to give steroid coverage during emergency situations for any patient taking steroids over the past 12 months, even if the patient took alternate-day glucocorticoids (Schlaghecke et al, 1992).
ADRENAL OVERACTIVITY
Cushing’s Disease and Cushing’s Syndrome
Cushing’s syndrome is a group of diseases characterized by excessive glucocorticoid secretion. The syndrome can be caused by increased ACTH secretion (pituitary or ectopic), or it can be ACTH-independent (adrenal disease). In Cushing’s disease, a subset of the group, a pituitary lesion leads to increased ACTH secretion, which in turn leads to bilateral adrenal hyperplasia and increased glucocorticoid production.
EPIDEMIOLOGY
The most common cause of systemic illness from exposure to excessive glucocorticoid is iatrogenic, from the use of exogenous glucocorticoids (steroids) to treat inflammatory or respiratory diseases. Endogenous excessive glucocorticoid production causing Cushing’s syndrome is most often ACTH-dependent. A pituitary tumor secreting excessive ACTH is the cause in 68% of all patients; this is called Cushing’s disease. Ectopic production of ACTH from a cancer or islet cell tumor occurs in 12% of cases and is also ACTH-dependent. ACTH-independent disease is caused by autonomous adrenal activity (adrenal adenomas or carcinomas) in 19% of cases (Orth, 1995). Cushing’s syndrome occurs at a rate of 10 cases per 1 million in the population.
HISTORY AND PHYSICAL EXAMINATION
Cushing’s syndrome should be considered in patients who have a history of hyperglycemia, hypertension, osteoporosis, and a recent gain in weight without use of steroids (Orth, 1995; Tsigos & Chrousos, 1994). The presence of marked virilization in women increases the likelihood of adrenal carcinoma, whereas marked hypertension may indicate ectopic ACTH secretion (Orth, 1995). The symptoms and signs of hypercortisolism are caused by alterations in the metabolism of lipids, protein, and carbohydrates. Signs and symptoms include:
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


