Cardiopulmonary Bypass
Charles A. Napolitano
Mauricio J. Palacios
Maria I. Castro
CASE SUMMARY
A 74-year old, 71 in., 82-kg man presents for a combined procedure: Aortic valve replacement, multivessel coronary artery bypass graft (CABG), and a left carotid endarterectomy (CEA). The patient has a history of longstanding hypertension, tobacco abuse, chronic obstructive pulmonary disease (COPD), diffuse coronary artery disease with 90% left main stenosis, moderate/severe aortic stenosis (51 mm Hg gradient; aortic valve area: 1.0 cm2), preserved left ventricular (LV) function (ejection fraction >50% and moderate LV hypertrophy), and peripheral vascular disease (severe left internal carotid artery stenosis and a 9-cm infrarenal abdominal aortic aneurism). Current medications include metoprolol, lisinopril, aspirin, and subcutaneous heparin. On the day of surgery, vital signs were: Pulse 67 bpm, blood pressure 101/63 mm Hg, respiratory rate 20 per minute, SpO2 97% to 98% with 2 L of O2 nasal cannula, hematocrit (Hct) 31%, and creatinine 1.6 mg per dL. Following induction with etomidate, cis-atracurium, and fentanyl, a right internal jugular central venous catheter (CVC) using ultrasonographic guidance and a transesophageal echocardiography (TEE) probe were placed. TEE examination confirmed the preoperative studies. The maintenance anesthetic was sevoflurane, O2-air mixture, cis-atracurium, and fentanyl. A bolus, followed by an infusion of aminocaproic acid was initiated following placement of the CVC. Surgically, an initial left CEA, utilizing procedural heparin and a shunt during carotid clamping, was followed by a 120-minute aortic crossclamp and a total cardiopulmonary bypass (CPB) time of 180 minutes for a four-vessel CABG with a left internal mammary artery (LIMA) to left anterior descending (LAD) artery, and the placement of a 23-mm porcine aortic valve. A seemingly uneventful intraoperative course that included the administration of 2 units of packed red blood cells (RBCs) to maintain Hct >25% during CPB moderate hypothermia (˜32°C) and a maximum warming temperature of 37°C during CPB, maintenance of “higher” pump flows and mean arterial pressure (MAP) between 60 to 80 mm Hg, 250 mL urine during CPB, preserved ventricular contractile function and a normal sinus rhythm with low dose infusions of nitroglycerin and dopamine following CPB, hemodynamic stability following heparin reversal with protamine, a dry surgical field before chest closure, and minimal chest tube drainage upon arrival in the ICU, was followed by a series of postoperative complications. Complications included left-sided weakness upon awakening, new onset atrial fibrillation on postoperative day (POD) 2, and an increased creatinine to 2.6 mg per dL.
Before discharge to a facility for continued rehabilitation of his cerebrovascular accident (CVA) symptoms, the patient’s creatinine returned to baseline and, following rate control intervention with a calcium channel blocker, atrial fibrillation resolved by POD 6.
▪ INTRODUCTION
Complications secondary to CPB are varied, as they occur in virtually every organ system for numerous reasons. Anticipated mechanical problems associated with initiation and/or maintenance of CPB (e.g., obstructed venous return, malpositioning of aortic cannula, air embolism, aortic dissection, oxygenator failure, arterial pump failure, and total electric failure) can yield catastrophic results. These anticipated problems can largely be avoided with vigilant observation and monitoring by the anesthesiologist, perfusionist, and surgeon. Their coordinated efforts are required for successful management of these intraoperative problems. The incidence, diagnosis, and management of these critical events during CPB have been reviewed thoroughly.1,2
Other complications that may not be specific to CPB or cardiac surgery, but reported to occur with similar incidences as other surgical procedures in the perioperative period, are described elsewhere in this monograph. Perhaps a noteworthy exception is that of injury to the ulnar nerve and brachial plexus3,4 where the incidence in cardiac surgery (2% to 38%) exceeds that of noncardiac surgery (0.02% to 0.06%). Apart from the controversy over ideal patient positioning, multiple mechanisms have been
postulated for increased incidence during cardiac surgery (e.g., excessive sternal retraction, internal mammary harvesting, duration of CPB and surgery, hypothermia, injury to the first rib or to the internal jugular vein). Again, vigilance on the part of the entire cardiac team with regard to patient positioning and to factors pertaining to the above-mentioned contributing mechanisms can be advocated to minimize this complication.
postulated for increased incidence during cardiac surgery (e.g., excessive sternal retraction, internal mammary harvesting, duration of CPB and surgery, hypothermia, injury to the first rib or to the internal jugular vein). Again, vigilance on the part of the entire cardiac team with regard to patient positioning and to factors pertaining to the above-mentioned contributing mechanisms can be advocated to minimize this complication.
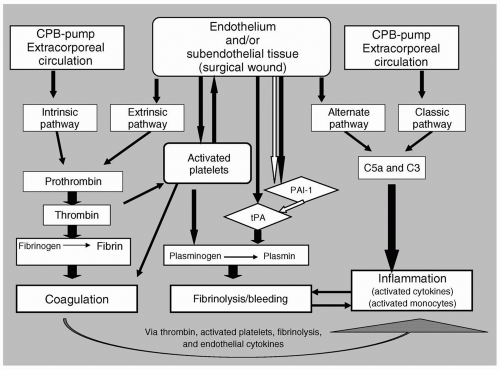 FIGURE 53.1 Schematic representation of activated coagulation, fibrinolytic, and inflammatory pathways in response to cardiopulmonary bypass(CPB). (Filled arrows represent stimulation; open arrows represent inhibition). PAI-1, plasminogen activator inhibitor 1; tPA, tissue plasminogen activator. |
Unpredictable or unexpected complications as a result of CPB, however, can result from the interplay of the patient’s pathophysiology and the normal physiologic response to CPB. This chapter focuses on the complications arising from the pathophysiologic responses to bypass. It is in this area that the anesthesiologist, by anticipating potential adverse events, may significantly impact patient management decisions. Therefore, a clear understanding of the physiologic response to CPB is required.
What Are the Hemostatic and Inflammatory Responses to Cardiopulmonary Bypass?
CPB exposes the circulating blood mass to extracorporeal circulation. The exposure of blood to foreign surfaces (e.g., biomaterials in the perfusion circuit), as well as to subendothelial tissues in the wound, activates both the intrinsic and extrinsic pathways of coagulation. The resulting thrombin formation and platelet activation produce a generalized “procoagulant” state. A reactive endothelium responding to input from circulating cytokines, having been generated by both hemostatic and inflammatory pathways, further enhances thrombin activation and platelet adhesion. CPB also activates and alters the fibrinolytic pathway. Under normal circumstances, this pathway is involved in limiting coagulation and promoting healing; however, during CPB, this pathway may be responsible for excessive blood loss. Lastly, the activation of coagulation and fibrinolytic cascades, as well as the direct contact with the perfusion circuit, serve to activate enzyme cascades and monocytes involved in inflammation. These inflammatory pathways may already be “primed” by the patient’s underlying pathophysiology, and in turn can exacerbate the activation of both thrombogenic and fibrinolytic pathways. The activation of thrombogenic, fibrinolytic, and inflammatory pathways does not occur in a linear manner, but rather, all these pathways appear to be stimulated simultaneously (see Fig. 53.1).
▪ COAGULATION CASCADE (EXTRINSIC VERSUS INTRINSIC)
The final step in the coagulation cascade is the formation of fibrin monomers from fibrinogen, which then
polymerize to form a fibrin clot. Activated thrombin serves as the enzymatic initiator of this final step and is itself formed by the activation of prothrombin by factor X in the presence of factor Va, phospholipid, and calcium.5 Two coagulation pathways, termed intrinsic (due to activation within the circulatory system) and extrinsic (due to activation from subendothelial collagen and thromboplastin), lead to a series of activated proteases that ultimately result in activated factor X and thrombin generation. Therefore, although the initiators of both intrinsic and extrinsic cascades differ, the result for both pathways is thrombin formation leading to production of a fibrin clot.
polymerize to form a fibrin clot. Activated thrombin serves as the enzymatic initiator of this final step and is itself formed by the activation of prothrombin by factor X in the presence of factor Va, phospholipid, and calcium.5 Two coagulation pathways, termed intrinsic (due to activation within the circulatory system) and extrinsic (due to activation from subendothelial collagen and thromboplastin), lead to a series of activated proteases that ultimately result in activated factor X and thrombin generation. Therefore, although the initiators of both intrinsic and extrinsic cascades differ, the result for both pathways is thrombin formation leading to production of a fibrin clot.
Traditional thought held that coagulopathies associated with CPB were mainly the result of intrinsic pathway activation through contact of the blood mass with the perfusion circuit.6 However, the extrinsic pathway is now recognized to have a greater clinical impact on hemostasis during CPB. Activation of this pathway plays a crucial role in the resulting coagulopathy and in activating a generalized inflammatory response that is typically seen in coronary artery surgical patients. When the initiating step (contact activation) of the intrinsic system is inhibited, thrombin and platelet activation still occur.7 Similarly, heparin-coated perfusion lines, which should block intrinsic pathway activation, minimally affect bleeding or thrombin generation during CPB.8 The use of off-pump coronary artery bypass (OPCAB) grafting has been advocated as a means to bypass the inflammatory and hemostatic response to CPB, as off-pump surgery completely eliminates the perfusion circuit. Some studies seem to indicate that OPCAB may result in improved clinical outcomes and a reduced need for blood transfusions.9 Other studies indicate that these improvements may be limited to the intraoperative periods as thrombin generation in the OPCAB group reaches that of the CPB group at the 20th to 96th hour of postoperative care.10 Global surgical trauma with both CABG and OPCAB seems to influence systemic inflammation and coagulation-fibrinolytic pathways, and may be as significant as the perfusion circuit in initiating the pathophysiologic changes in hemostasis associated with heart surgery.11 Lo et al. measured the activation of hemostasis in 20 off-pump and 20 on-pump coronary artery surgical patients.10 Total thrombin formation, as measured by the generation of prothrombin fragment 1.2 during and following coronary artery surgery, was significantly greater in on-pump patients than off-pump patients, rising 450% above baseline in on-pump patients as compared to 342% above baseline in off-pump patients. The pattern of thrombin generation showed differences with the on-pump group generating peak thrombin levels immediately after surgery that remained elevated for four subsequent days and the off-pump group generating gradual increases in thrombin that had similar peak values at day 4 as the on-pump group. The presence of D-dimers (fibrinolytic activation) and von Willebrand factor (vWF) (measuring endothelial activation) rose to a similar extent in both groups. Therefore, the removal of intrinsic pathway activation with OPCAB does not completely remove thrombin generation in response to surgery, nor does it prevent fibrinolytic or endothelial activation.
▪ FIBRINOLYTIC PATHWAYS
Fibrinolysis occurs by the action of plasmin on the fibrin clot to form degradation products that include D-dimers. Plasmin is generated during CPB by action of the tissue plasminogen activator (tPA) on the inactive enzyme, plasminogen. Plasma levels of tPA increase within 30 minutes of CPB and return to baseline by the first POD.12 Therefore tPA release, plasmin formation, and consequent fibrinolysis occur simultaneously with thrombin formation and coagulation. Endothelial cells release tPA in response to kallikrein, an activated component of the complement cascade, and thrombin. β-agonists, bradykinin, and components of platelet activation can also act upon endothelial cells to promote tPA release. The tPA is itself inhibited by antiplasmin (C1 inhibitor) and plasminogen activator inhibitor 1 (PAI-1) released by the endothelium.13
PAI-1, by its action on tPA, plays a key regulatory function in fibrinolysis: Low levels of PAI-1 can tip hemostasis toward increased fibrinolysis and bleeding, whereas high levels of PAI-1 will, by virtue of inhibiting tPA and subsequent plasmin formation, tip hemostasis toward coagulation. Plasma PAI-1 levels also rise in response to inflammatory signals. PAI-1 is not stored in endothelial cells; its release is the product of upregulation of de novo synthesis. Although endothelial cells are stimulated early in the bypass procedure to upregulate PAI-1 production, increases in PAI-1 are seen toward the end of CPB, and peak 12 to 36 hours post CPB. Therefore, increased PAI-1 levels put a patient at risk for postoperative thrombosis. Increased basal levels of PAI-1 are seen in patients with insulin resistance syndrome. Various polymorphisms for PAI-1 exist; the 4G allele polymorphism results in higher levels of PAI-1 in response to various stimuli.13,14 Those individuals with the 4G allele genotype may be more at risk for perioperative thrombosis during and following CPB. Conversely, those individuals with deficits in PAI-1 production may be at higher risk for perioperative hemorrhage.
▪ PLATELET ACTIVATION
Platelet activation, dysfunction, and subsequent loss have long been a documented consequence of CPB.14,15,16,17 Hemodilution, adhesion, aggregation, release, and destruction reduce platelet number by 30% to 50%.15,16 It is important to remember that platelet-endothelial interactions and platelet activation is an immediate first response to bleeding in the high flow and high pressure arterial side of the vasculature. Circulating thrombin also plays a major role in platelet activation, albeit on the slower venous side of the circulation.17 Therefore, signals that would normally serve as physiologic triggers to prevent blood loss become magnified during CPB and result in general platelet depletion.
Normally, platelets exist in a “quiescent” state due to the presence of anticoagulant factors from the epithelium. On the arterial side, high flow rates dilute and disperse cells and coagulation factors. The secretion of nitric oxide (NO) and prostacyclin from the epithelium serve not only
to maintain a vasodilated state but also to inhibit platelet activation. Endothelial production of heparin sulfate, thrombomodulin, and protein C also act as endogenous anticoagulant factors on the venous side by their action on platelet adhesion/activation, activation of protein C, and cleavage of coagulation factors Va and VIIIa, respectively. An intact epithelium is therefore required for normal platelet action on both venous and arterial sides of the vasculature.
to maintain a vasodilated state but also to inhibit platelet activation. Endothelial production of heparin sulfate, thrombomodulin, and protein C also act as endogenous anticoagulant factors on the venous side by their action on platelet adhesion/activation, activation of protein C, and cleavage of coagulation factors Va and VIIIa, respectively. An intact epithelium is therefore required for normal platelet action on both venous and arterial sides of the vasculature.
During damage or trauma, exposure of platelets to arterial subendothelial collagen and vWF causes rapid activation of platelets and adhesion on endothelial surfaces because of low affinity binding between vWF monomers and glycoprotein I (GPI) of platelet membranes. This binding causes the platelet to roll along the endothelial surface and further activate GPII and GPIII proteins on the platelet surface to firmly bind the platelet to vWF multimers and fibrinogen. Activated platelets also provide a negatively charged phospholipid surface on which coagulation reactions can occur and receptors for coagulation factors Xa, IXa, and Va. On the venous side, the circulating thrombin generated through both extrinsic and intrinsic coagulation may be the most likely stimulus to activate platelets. Heparin increases the sensitivity of platelets to soluble agonists, inhibits binding to vWF, and modestly increases template bleeding times. As CPB continues, activated complement, plasmin, hypothermia, platelet activating factor, interleukin 6 (IL-6), cathepsin G, serotonin, epinephrine and other agonists also activate platelets and contribute to their loss and dysfunction. Overall, the function of the platelet mass is reduced, and bleeding times are prolonged to approximately twicenormal values for several hours following the conclusion of CPB and heparin-protamine binding.15
▪ EFFECTS OF ENDOTHELIUM
The serum enzymes involved in the thrombogenic and fibrinolytic cascade pathways do not exist in isolation; they are constantly exposed to and interact with reactive peptides involved in inflammation, with platelets and other blood cells, and with the endothelium. Under normal conditions, the endothelium plays a protective role by containing and secreting endogenous anticoagulants, such as thrombomodulin, tPA, and heparin-like molecules. The endothelium also protects against high velocity stress in small arterioles by producing “relaxing factors”, namely NO and prostacyclin. Therefore, under normal circumstances, the endothelium plays a protective and limiting role for thrombogenesis. What then is altered in the cardiac patient undergoing CPB for coronary artery graft or valve surgery?
Diffuse endothelial activation or damage occurs during CPB and cardiac surgery, and has probably already occurred as a result of preexisting disease. During injury, the expression of endothelial anticoagulant proteins is downregulated and that of procoagulant peptides are upregulated.7 Upon injury, endothelial cells lose their tight connectivity, allowing serum proteins to be exposed to subendothelial connective tissue. Finally, injury results in apoptosis of endothelium, the dead cells then becoming chemotactic for platelets and procoagulant. Evidence for all these endothelial changes has been observed in experimental models of CPB and may eventually explain the variety of responses of patients to similar stimuli.
Endothelial cells are also activated by cytokines involved with inflammation, including C5a, IL-1, and tumor necrosis factor (TNF).18,19 Epithelial activation through inflammatory mediators triggers the endothelium to express procoagulant and fibrinolytic proteins and also activates the endothelial expression of membrane adhesion molecules specific to the adhesion and eventual transmigration of neutrophils and monocytes.20
Endothelial cells are sensitive to hypoxia, temperature changes, circulating cytokines, endotoxin, cholesterol, nicotine, surgical manipulation, and hemodynamic sheer stress. Events related to CPB, the surgical process itself, or reperfusion of previously injured tissue can therefore globally stimulate endothelium to become more procoagulant. Preexisting influences such as hypertension, smoking, diabetes, or hyperlipidemia also disrupt endothelial cells. Therefore the hypertensive patient with a history of smoking, hyperlipidemia, and type 2 diabetes probably enters the operating suite with an activated endothelium that, when exposed to perturbations resulting from cardiac surgery and/or CPB, can result in marked global activation of thrombin formation, platelet consumption, and activation of fibrinolytic pathways.7
▪ INFLAMMATION
Inflammatory cascades involve complement activation by both the classic and alternative pathways.20 The classic pathway is activated by the surface contact in the perfusion system or by antigen-antibody complexes. The alternative pathway also forms C3 convertase, and is activated by polysaccharides from bacteria or other cell membranes; it is actually the predominant pathway mediating inflammation following CPB. Both pathways culminate in the activation of complement factor C5a. Factor C5a, a potent chemotactic agent that initiates leukocyte aggregation and activation, has been implicated in mediating the systemic inflammatory response syndrome (SIRS) associated with CPB. At the end of CPB, the classic pathway is activated a second time by the heparin-protamine complex.21 Increased C3a and C5a levels seem to be associated with cardiac and pulmonary dysfunction.
The activation of complement, coagulation, fibrinolysis, and platelets also activates blood cells involved in mediating an inflammatory response.21 During CPB, neutrophils release neutral proteases, lysosomal enzymes, myeloperoxidase, hydrogen peroxide, hydroxyl radicals, hypochlorous acid, acid hydrolases, and collagenases.21 These compounds amplify further inflammatory or coagulation cascades and white blood cell activation. Neutrophils in this scheme play a major role in ischemiareperfusion injuries and are responsible for much of the inflammatory response associated with CPB.20 Monocyte activation during CPB produces the release of various cytokines (including IL-1, IL-6, and IL-8) and TNF. The
resulting activation of an inflammatory response has been implicated in end-organ injury or dysfunction. Furthermore, the number of B and T lymphocytes and the responsiveness of lymphocytes to mitogens and other agonists are reduced during the first week after CPB, thereby increasing the likelihood of infection.21,22
resulting activation of an inflammatory response has been implicated in end-organ injury or dysfunction. Furthermore, the number of B and T lymphocytes and the responsiveness of lymphocytes to mitogens and other agonists are reduced during the first week after CPB, thereby increasing the likelihood of infection.21,22
▪ ISCHEMIC-REPERFUSION INJURY
Diffuse arteriosclerosis and/or coronary artery disease set up conditions of limited flow and O2 delivery to end organs with a potential for ischemic injury. The reperfusion of ischemic tissue following aortic crossclamping, nonpulsatile CPB, and/or resultant coronary artery reanastomoses delivers platelets, inflammatory blood cells and enzymes involved in both inflammatory and hemostatic cascades to tissue or endothelium that is already in a primed or proinflammatory state. Subsequent activation of inflammation, with ensuing release of vasoactive cytokines, produces microcirculatory vasodilation that manifests itself as hypotension and hypoperfusion of end organs.22
Short periods of ischemia (i.e., preconditioning) have been shown to provide a protective effect against subsequent longer ischemic periods in a variety of organs, including the heart, liver, lung, kidney, and brain. Ischemic preconditioning is a cellular response to injury that is mediated by adenosine and NO. ATP-sensitive potassium (K+-ATP) channels in cellular and mitochondrial membranes are important in mediating the effects of preconditioning. Blockade of these ion channels in animal studies of reperfusion injury negate the protective effects of preconditioning.23
What Are the Current Strategies to Minimize Coagulation, Bleeding, and Systemic Inflammation Associated with CPB?
▪ COAGULATION
Heparin is the drug of choice for anticoagulation during extracorporeal circulation because of its immediate onset of action, its reversibility, and its ability to inhibit coagulation throughout the CPB circuit.24,25 Heparin acts by binding to antithrombin III and thereby accelerates antithrombin III binding to thrombin.25 The goal of providing adequate anticoagulation is to overwhelm the system with high doses of heparin, causing a putative sink for the antithrombin III-thrombin complexes. Heparin also has direct effects on platelets and other enzymes in the coagulation and inflammatory pathways. Furthermore, heparin inhibits coagulation at the end of the cascade after most coagulation factors have been converted to active enzymes that can influence neutrophil and monocyte activation or inflammatory cascades. Therefore, many of the hemostatic or inflammatory complications attributed to CPB could also be due to a direct effect of heparin and/or heparin-protamine complexes.24,25
In certain individuals, heparin can be associated with an allergic response, which can range from mast cell release of histamine following bolus doses to devastating heparin-induced thrombocytopenia (HIT). In those patients who are prone to HIT, various other anticoagulant regimens have been used.26,27,28,29,30 The use of direct thrombin inhibitors has produced acceptable outcomes in cardiac surgery with and without CPB. Their anticoagulant effect, which can be monitored by activated clotting time (ACT) and/or activated partial thromboplastin time (aPTT), occurs immediately and, although these compounds have no established antidote, they have relatively short halflives (˜30 to 60 minutes). Hemofiltration can hasten the anticoagulant reversal. The direct thrombin inhibitors currently used clinically include recombinant-hirudin, Argatroban, a synthetic L-arginine, and Bivalirudin, a semisynthetic hirulog. Low molecular weight heparins (nadroparin, dalteparin, and enoxaparin) are other alternatives to heparin.29,30 Their anticoagulative effect is due to conformational changes of antithrombin and heparin cofactor II.
Low molecular weight heparins affect factor IIa (thrombin) much less than factor Xa compared to unfractionated heparin. The indications for this strategy in CPB are currently unclear because cross-reactivity in patients with HIT has been reported to be up to 90%, the half-life of these molecules is longer than that of heparin, and there are no coagulation assays to monitor their activity.
Regarding the CPB circuit, the strategy is to create surfaces that will not activate the intrinsic system. The use of surface-bound heparin (bound with either covalent or ionic bonds) is one approach. Others have employed circuits with surface-modifying additives (SMA), aimed at reducing contact activation. These SMA are copolymers that can be either added directly to the base polymer resins before processing the circuit components or coated onto the blood-contact surfaces. A recent clinical trial by Defraigne et al. showed that the platelet count in the SMA group decreased less, and there was a reduction in the thromboglobulin plasma level. The decrease in blood loss, however, did not reach a statistically significant level, except in the group of patients taking aspirin preoperatively.31,32
▪ PLATELET PRESERVATION
Currently, the mainstay strategy to deal with platelet loss and dysfunction during CPB is platelet transfusion. A newer strategy that focuses on the preservation of platelets is known as platelet anesthesia.33,34 Platelet activation during CPB can be suppressed by agents such as tirofiban and eptifibatide, which are inhibitors of platelet
aggregation. These two agents inhibit platelet aggregation by reversibly binding to the platelet receptor GP IIb/IIIa, thereby preventing the binding of fibrinogen. Inhibition of platelet aggregation occurs in a dose- and concentration-dependent manner. These drugs are administered in combination with heparin to prevent ischemic complications in patients with acute coronary syndrome. Their use in platelet preservation during cardiac surgery is a novel approach and one awaiting clinical outcome trials. Both tirofiban and eptifibatide, when used in combination with NO, showed successful preservation of platelets in primates.33,34 Using NO alone for this purpose showed partial protection.35 NO stimulates guanylate cyclase to increase the levels of cyclic guanosine 3′, 5′ monophosphate (cGMP), which inhibits platelet aggregation and adhesion, as well as cGMP-inhibited cyclic adenosine 3′, 5′ monophosphate(CAMP) phosphodiesterase activity. Inhibition of this phosphodiesterase increases platelet cAMP, which decreases cytosolic calcium ion and inhibits basic platelet reactions (e.g., aggregation, adhesion, secretion of dense and α-granule contents, and release of acid hydrolases).34 Since NO and platelet aggregation inhibitors work through different mechanisms, a synergistic effect may be expected. However, preliminary studies in primates have not shown this synergistic effect. Regarding the reversal of effects, the actions of NO will dissipate upon its termination, whereas the <2-hour half-life of the platelet aggregation inhibitors will coincide with the duration of most CPB procedures. Preliminary studies report the return of normal bleeding times 30 to 60 minutes after the administration of protamine.33,34
aggregation. These two agents inhibit platelet aggregation by reversibly binding to the platelet receptor GP IIb/IIIa, thereby preventing the binding of fibrinogen. Inhibition of platelet aggregation occurs in a dose- and concentration-dependent manner. These drugs are administered in combination with heparin to prevent ischemic complications in patients with acute coronary syndrome. Their use in platelet preservation during cardiac surgery is a novel approach and one awaiting clinical outcome trials. Both tirofiban and eptifibatide, when used in combination with NO, showed successful preservation of platelets in primates.33,34 Using NO alone for this purpose showed partial protection.35 NO stimulates guanylate cyclase to increase the levels of cyclic guanosine 3′, 5′ monophosphate (cGMP), which inhibits platelet aggregation and adhesion, as well as cGMP-inhibited cyclic adenosine 3′, 5′ monophosphate(CAMP) phosphodiesterase activity. Inhibition of this phosphodiesterase increases platelet cAMP, which decreases cytosolic calcium ion and inhibits basic platelet reactions (e.g., aggregation, adhesion, secretion of dense and α-granule contents, and release of acid hydrolases).34 Since NO and platelet aggregation inhibitors work through different mechanisms, a synergistic effect may be expected. However, preliminary studies in primates have not shown this synergistic effect. Regarding the reversal of effects, the actions of NO will dissipate upon its termination, whereas the <2-hour half-life of the platelet aggregation inhibitors will coincide with the duration of most CPB procedures. Preliminary studies report the return of normal bleeding times 30 to 60 minutes after the administration of protamine.33,34
▪ BLEEDING
Exclusive of good surgical technique, perioperative blood loss has mainly been controlled by the antifibrinolytics tranexamic acid (TXA), ε-aminocaproic acid (EACA), and aprotinin.13,36 EACA and TXA are both synthetic lysine analogs that inhibit plasmin-related fibrinolysis and plasminogen binding to fibrin with subsequent activation to plasmin.13 Aprotinin, a nonspecific serine protease inhibitor, affects fibrinolysis at multiple points. Aprotinin inhibits tPA release by competing with plasmin for receptor binding sites and by inhibiting kallikrein activation. Other factors that may contribute to decreased blood loss include inhibition of serine proteases involved in the clotting cascade and preservation of platelet function.13 Additionally, aprotinin has been shown to exert anti-inflammatory properties by the inhibition of enzymes involved in inflammatory cascades. Therefore, because of its multiple sites of action, aprotinin gained popularity as an agent of choice for patients at high risk for postoperative bleeding. In the late 1980s, with the rising cost of blood transfusion and the increasing risk of acquired infection from allogenic blood (e.g., human immunodeficiency virus [HIV], hepatitis C), the established efficacy of aprotinin prompted its use in a wider range of patients.
While aprotinin’s efficacy in controlling blood loss is unquestioned,13 controversy exists as to its safety profile.37 Initial premarketing clinical trials with aprotinin did not report significant adverse events, except for the possibility of anaphylactic reactions to repeated doses, which were manifested mainly as severe hypotension.38,39 Two postmarketing observational studies have reported adverse outcomes associated with aprotinin use, mainly renal dysfunction.40,41 Both studies used propensity statistical scores to adjust for differences between patient groups (i.e., preexisting disease profiles). The reasons for the aprotinin-associated increase in renal dysfunction are not well understood; it is possible that the antifibrinolytic action favors formation of microemboli, which then are responsible for end-organ ischemia and damage, or that aprotinin directly inhibits formation of protective cytokines at the end-organ level. Similarly, while the studies by Mangano et al. and Karouti et al. seem to indicate that TXA or EACA are safer alternatives in terms of end-organ damage, this also remains to be definitely demonstrated.40,41
Although it is possible that these observational studies, by their very nature of not being randomized, double-blinded controlled studies, could have resulted in a disproportionate number of patients at risk for renal failure from other causes in aprotinin treatment groups.40,41 It is also possible that premarketing clinical trials, given their strict inclusion and exclusion criteria, could have resulted in the omission of patients at risk for developing renal complications. A retrospective review of 67,000 patients also suggests that aprotinin is associated with increased death, renal failure, heart failure, and stroke.38,39 In light of these new studies, the U.S. Food and Drug Administration (FDA) modified its prescribing information with more focused indications, more warnings about adverse events, and new contraindications.39 As of December 15, 2006, recommendations and considerations for physicians include the following:
Aprotinin use should be limited to only those patients at increased risk for blood loss and blood transfusion in association with CPB in the course of coronary artery bypass grafting. Aprotinin should be limited to those situations where the clinical benefit of blood loss is essential to the medical management of the patients, and the benefit outweighs the risk.
Patients should be carefully monitored for the occurrence of toxicity, particularly of the kidney, heart, or central nervous system or for signs of anaphylactic reactions. Physicians should be aware of new product labeling that warns of increased risk for renal dysfunction, potential increase in postoperative dialysis, stronger warnings regarding anaphylactic reactions to aprotinin, and a contradicted use in patients with prior exposure to aprotinin or aprotinin-containing products (i.e., fibrin glues such as Tissucol or Tisseel). (Tissucol or Tisseel, Baxter International, Deerfield, IL).
Serious and unexpected adverse events should be reported to the drug manufacturer and/or the FDA MedWatch program.
Patient counseling should include the indications for use of aprotinin (e.g., high risk of intraoperative bleeding), risks associated with aprotinin (e.g., anaphylaxis and renal dysfunction), and preexisting factors that increase a patient’s chance for developing complications
in association with aprotinin use (e.g., prior heart surgery, prior treatment with aprotinin, known drug allergies, known kidney disease).
The more recent multicenter, observational study by Mangano et al.37 which reviewed the 5-year mortality after CABG surgery in more than 1,000 patients reveals an expected versus observed 5-year mortality in aprotinintreated patients of 1.48. This excess mortality was not found in aminocaproic- or TXA-treated patients whose 5-year mortality was similar to control.37 The authors conclude that the use of aprotinin does not seem prudent given the excess mortality and safer alternatives.
▪ INFLAMMATION/REPERFUSION INJURY
Hypotension and low organ perfusion is a result of vasodilation associated with the systemic inflammatory response. Maintenance of cardiac output (CO) and endorgan perfusion pressure is accomplished by the intravascular administration of fluids, inotropes, and other pharmacologic agents. Ischemia produces a proinflammatory state in end organs that can be attenuated by preconditioning. Treatment modalities that attenuate this inflammatory response or mimic preconditioning are being pursued as adjuncts to traditional therapy. Adenosine is a mediator in preconditioning, and infusions of adenosine or adenosine-1 receptor agonists have been advocated as interventions to ischemic reperfusion injury. Opiates, particularly morphine, seem to mimic preconditioning in in vitro and in vivo models. Inhalation agents such as isoflurane also can mimic preconditioning effects. Therefore, the effects of reperfusion injury may be attenuated by deliberately choosing specific anesthetic and analgesic agents.23
Therapeutic approaches to attenuate systemic inflammation have included aprotinin, steroids, and inhibition of complement protein C5. Aprotinin, as discussed previously, affects inflammation by inhibiting proteases involved in the complement cascade.7,42 The effects of corticosteriods on SIRS in cardiac surgical patients remains unsettled.7,43 Studies have shown that glucocorticoids blunt the release of cytokines involved in inflammation (e.g., TNF, IL-6, IL-8) leading to improved hemodynamic stability. However, glucocorticoid therapy results in immunosuppression and hyperglycemia that may exacerbate postoperative complications. Preclinical animal studies of pexelizumab, a monoclonal antibody against complement protein C5, have shown that pexelizumab decreases the amount of myocardial damage associated with ischemia and reperfusion.44 Despite success with C5 antibody therapy in myocardial infarction (MI) patients, clinical trials in CABG patients have been equivocal. Phase II trials of pexelizumab bolus plus infusion to patients undergoing CABG or CABG/aortic valve replacement (AVR) showed an improvement in primary outcomes at 4 or 30 days (MI and/or mortality), but not at 90 days in CABG-only patients.45 While pexelizumab also did not improve the overall incidence of neurocognitive damage, it did significantly improve tasks involving the visual-spatial domain.46 A phase III trial of 3,099 patients in 205 centers showed that, although pexelizumab only had a tendency to decrease mortality by 18% at 30 days in CABG-only patients, it significantly decreased mortality or postoperative MI in those patients undergoing CABG plus AVR. All patients were considered “high risk” and had at least one of the following risk factors: Diabetes, prior CABG, urgent need for surgery, female gender, prior history of neurologic event, prior history of congestive heart failure (CHF), prior history of more than two MI or recent MI within the last 30 days. Pexelizumab decreased mortality by 28% in those patients with two or more concomitant risk factors (67% of the total patient population).47 These results seem to indicate the following: The morbidity associated with CPB is multifactorial; anti-inflammatory treatment may benefit some groups of patients more than others; and multiple interventions may be needed to address the complex pathophysiologic response to CPB.
▪ ON-PUMP VERSUS OFF-PUMP
During CPB, venous blood from the right auricle drains by gravity into an external reservoir and is then pumped through an oxygenator back into the systemic circulation generally through a cannula placed in the ascending aorta. By placing a clamp across the aorta proximal to the cannula, the heart and lungs are effectively removed from the circulation. Because the CPB apparatus assumes the functions of the heart and lungs, the heart is emptied and rendered electrically and mechanically silent with cardioplegia. CPB entails the extracorporeal transit of blood, nonpulsatile blood flow, hemodilution, and some degree of hypothermia.
OPCAB has been proposed as a means by which the proinflammatory and procoagulant effects of the bypass circuit can be circumvented. OPCAB reduces the inflammatory response associated with CPB22 but does not affect other inflammatory stimuli such as surgical trauma, reperfusion injury, and thrombin activation. Studies comparing OPCAB with CPB-assisted CABG have shown modest reductions in coagulation or inflammation with OPCAB. Conflicting data exist regarding the effects of OPCAB on complement activation or IL-6 production. OPCAB seems to reduce the generation of circulating IL-8, a potent stimulus for neutrophil migration and myocardial ischemic injury. Outcome measures of death, stroke, or MI suggest a trend toward the beneficial effect of OPCAB, though meta-analysis of comparable trials has not shown a significant effect of OPCAB when compared with CPB.7 Other analyses of OPCAB seem to indicate reduced morbidity and mortality associated with reduced bleeding, making OPCAB a viable option for those patients at high risk for perioperative bleeding or in whom blood transfusions are not an option (e.g., Jehovah’s Witnesses).48 The beneficial effect of OPCAB on end-organ function is still being determined. It is
evident that complications resulting from CABG are multifactorial and not only because of the effects of extracorporeal perfusion. Table 53.1 illustrates several of the differences and similarities of OPCAB and CABG with CPB (on-pump). Although extracorporeal circulation, along with its concomitant physical characteristics, is eliminated with the OPCAB technique, several of the currently used protocols (e.g., heparin and protamine administration, aortic and beating heart manipulation) remain in place. These similarities of technique may obscure identification of additional benefits from the avoidance of CPB.
evident that complications resulting from CABG are multifactorial and not only because of the effects of extracorporeal perfusion. Table 53.1 illustrates several of the differences and similarities of OPCAB and CABG with CPB (on-pump). Although extracorporeal circulation, along with its concomitant physical characteristics, is eliminated with the OPCAB technique, several of the currently used protocols (e.g., heparin and protamine administration, aortic and beating heart manipulation) remain in place. These similarities of technique may obscure identification of additional benefits from the avoidance of CPB.
TABLE 53.1 Coronary Artery Bypass Graft Surgery with Cardiopulmonary Bypass (CPB)(On-Pump) versus Coronary Artery Bypass Surgery (Off-Pump) | |||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
▪ TIGHT GLYCEMIC CONTROL
Hyperglycemia, a common occurrence during CPB in both nondiabetic as well as diabetic patients, is associated with adverse outcomes including: increased length of intensive care unit (ICU)/hospital stay, increased incidence of dysrhythmias, depressed myocardial contractile function, recurrent ischemia, need for postoperative inotropic support, increased incidence of wound infection, and increased mortality.49,50,51,52,53,54,55 Tight glycemic control has been associated with a decreased incidence in the above outcomes. Surprisingly, tight glucose control has failed to improve neurologic or neurobehavioral outcomes in patients without diabetes mellitus undergoing CABG.55 Whether the etiology of hyperglycemia is due to the stress response alone or in conjunction with the release of proinflammatory cytokines, catecholamines, glucagon, growth hormone, and glucocorticoids, or is the result of insulin resistance in the diabetic or “prediabetic” patient, or the influence of various cardioplegia solutions containing dextrose, hyperglycemia has been shown to impact cellular components regulating metabolism,51 immune cell activation and circulating cytokines,56 and myocardial ion channels.57 Clinical studies addressing diabetic cardiac surgical patients have suggested that tight glucose control (125 to 200 mg per dL) in the perioperative period is optimal and results in a lower incidence of sternal wound infections and postoperative mortality.51 Nondiabetic patients who are exposed to the stress of cardiac surgery have also been shown to benefit from tight glycemic control.53 The direct relation between fasting glucose levels and the risk of sustaining a cardiovascular event and the relation between admission blood glucose levels following an acute MI and long-term mortality has established hyperglycemia as an independent predictor of cardiovascular risk.51,57 These studies have also established the benefit of aggressive treatment with insulin and resultant tight glucose control.58 On the basis of these clinical studies, a reasonable recommendation for perioperative glycemic control is maintenance of blood glucose at ≤150 mg per dL before CPB and at ≤180 mg per dL during CPB.58 Initiation of an insulin drip in the operating room is a very useful tool to help effect and maintain tight perioperative glucose control.
Mechanisms for improved outcomes following tight glycemic control have been suggested to be the result of an attenuated inflammatory response, decreased endothelial injury, decreased circulating levels of noxious factors including cytokines and free O2 radicals, and/or decreased metabolic demand on reperfused tissue. Acute and chronic hyperglycemia reduces the “preconditioning” effects of anesthetics through mechanisms involving the inhibition of potassium channels that regulate vascular perfusion and/or myocyte contractility. Sulfonurea-based oral hypoglycemic agents, by virtue of their action on potassium channels, also seem to further negate preconditioning.59 On the basis of these observations and inferences, it may be prudent to withhold sulfonureabased hypoglycemic agents (i.e., glyburide) 24 to 72 hours before cardiac surgery and, if appropriate, initiate insulin therapy. Metformin, an oral hypoglycemic agent that lowers glucose levels by sensitizing target tissues to insulin, inhibits hepatic glucose production, and increases peripheral glucose uptake, may be an appropriate agent to continue in the perioperative period.58 The interplay of the many cellular and humoral systems with glucose and preexisting pathophysiology remains an active area of investigation.
What Are Common End-Organ Complications of CPB?
The pathophysiology associated with coronary artery disease, when coupled with other comorbid disease states (e.g., hypertension, diabetes) and the activation of hemostatic and inflammatory cascades, makes every organ in the body susceptible to organ dysfunction. The most common and serious complications of CPB occur in the brain, heart, kidneys, and lung. Gastrointestinal (GI) complications, although not common, are associated with a high mortality rate.
▪ NEUROLOGIC COMPLICATIONS
The patient undergoing CPB is at significant risk of developing neurologic complications. These complications can be divided into frank stroke, encephalopathy, and neurocognitive disorders. The typically reported frequency of stroke varies from 1% to 6%. Valve surgery, if combined with other preexisting factors, may increase this incidence to 9%.60 The Multicenter Study of the Perioperative Ischemia (McSPI) Research Group and the Ischemia Research and Education Foundation (IREF) has classified all neurologic injury into type 1 and type 2 with independent risk factors61 (see Table 53.2). Patients undergoing combined CABG and open ventricle procedures have a higher incidence of neurologic complications, reaching almost 16%. The patient case presented at the beginning of this chapter describes a patient who was at risk for developing neurologic injury following CPB and surgery. A stroke risk index to predict neurologic complications following CABG is under development.60,62
The etiology of these neurologic injuries is multifactorial and can include embolization, hypoperfusion, and/or ischemic/reperfusion injury. Probably the one most important factor for neurologic injury is embolization of brain areas. Transcranial Doppler, retinal angiography, pathology, and radiology have served to document micro- and macroembolisms during and after cardiac surgery.63,64 It is probable that all patients experience some degree of embolization during CPB.64,65 The central nervous system is exposed to hundreds of embolic events during bypass that can result in transient or permanent ischemia. Ischemia resulting from brain hypoperfusion is another factor implicated as a cause of morbidity related to neurologic adverse outcomes.66,67 Although MAP during CPB has not been proven to be a primary determinant of adverse neurologic outcome, regional brain hypoperfusion probably contributes as a result of preexisting vascular pathology secondary to hypertension, diabetes, or senile atherosclerotic disease.68,69,70 The fact that tight glycemic control does not affect neurologic outcomes argues in favor of preexisting microcirculatory damage being exacerbated or initiated during CPB in the diabetic patient.55 Finally, the role by which inflammation causes ischemic injury in the brain is an area of active research. It is becoming clear that the interactions between inflammatory mediators, hypoperfusion, and the endothelium are important in determining subsequent damage. Several questions regarding the interplay between inflammation, ischemia, and neurologic injury still need to be resolved. It is possible that the inflammatory response triggered by CPB converts otherwise trivial ischemic insults into clinically important pathology.60 It is not clear whether CPB-related inflammation is enough to alter endothelial function in the absence of cerebral ischemia.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


