Talmage D. Egan
I. Brief History and Overview
Analgesics are an essential resource for the provision of anesthesia and treatment of postoperative pain. Both opioid and nonopioid analgesics will be reviewed in this chapter. Opioid analgesics predate modern medicine while nonsteroidal anti-inflammatory drugs (NSAIDs), with their anti-inflammatory, antipyretic, and analgesic qualities, have become integral parts of perioperative medicine in the past 30 years.
The first NSAIDs to be isolated were salicylic acid derivatives. Traditional NSAIDs inhibit prostanoid synthesis through nonspecific inhibition of the cyclooxygenase (COX) enzymes. Prostanoids are produced in response to tissue injury and inflammation, both peripherally and centrally. This contributes to peripheral pain sensitization, general pain perception, and the sickness syndrome consisting of fever, anorexia, and changes in mood and sleep patterns (1). Prostanoids are also involved in homeostatic functions in the kidneys, gastric mucosa, platelets, and central nervous system (CNS). They are derived from arachidonic acid, which is released from cell membranes during tissue injury and inflammation, and consist of prostaglandins, including prostacyclin, and thromboxane A2. Multiple enzymes are involved in the transformation of arachidonic acid into prostaglandins and thromboxane A2, including isoforms of the COX enzymes (Fig. 10-1). The characterization of the COX-2 isoform as “inducible” (by neurotransmitters, growth factors, and proinflammatory cytokines) and the COX-1 isoform as “constitutive” is based on the predominant function of each isoform. But this is an oversimplification and both isoforms have some constitutive and some inducible functions. The COX-3 isoform remains poorly understood. Although aspirin irreversibly inhibits cyclooxygenase and thus platelet aggregation (see discussion below), the other NSAIDs reversibly inhibit cyclooxygenase so that enzymatic function is restored as the drug is cleared from the circulation, leading to a dose-dependent and drug-dependent effect on platelet aggregation.

Aspirin irreversibly inhibits platelet aggregation for the life of the platelet, while other non-COX-2 specific, nonsteroidal anti-inflammatories reversibly inhibit aggregation in a dose-dependent manner until the drug is cleared from the circulation.
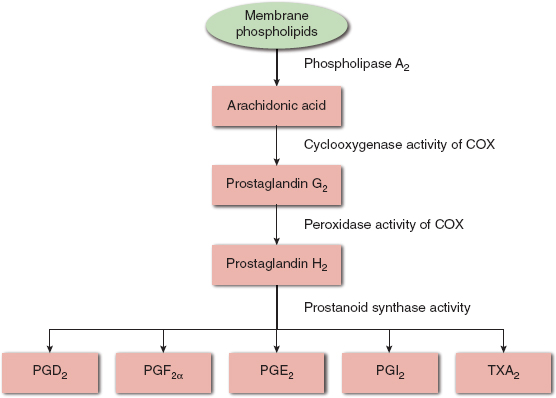
FIGURE 10-1 Schematic view of prostanoid synthesis. COX, cyclooxygenase enzymes; PG, prostaglandin; TX, thromboxane; PGD2, prostaglandin D2; PGF2α, prostaglandin F2α; PGE2, prostaglandin E2; PGI2, prostaglandin I2; TXA2, thromboxane A2.
II. Nonopioid Analgesics
A. Acetaminophen
Indications, Contraindications, and Dose
Acetaminophen (also known as paracetamol, APAP, para-acetylaminophenol, N-acetyl-para-aminophenol) is widely used as an analgesic and antipyretic for both children and adults. Although it has been reported to suppress inflammation in animals and in dental tissue, the anti-inflammatory effect is considered low to nonexistent (2). Centrilobular hepatotoxicity occurs with acute overdose and may occur at smaller doses in chronic alcoholics, although the relation between acetaminophen use and alcohol ingestion is not straightforward. Use of acetaminophen in patients with chronic liver disease who do not regularly consume alcohol is not contraindicated, although the maximum safe dose has not been determined (3). It is available in oral, rectal, and intravenous preparations. The dose is 10 to 15 mg/kg every 4 to 6 hours in children (every 6 to 8 hours in neonates) and 325 to 650 mg every 4 to 6 hours in adults. The maximum dose in children is 75 mg/kg/day and, historically, no more than 4 g/day in adults. However, due to the availability of combination products (over-the-counter cough and cold preparations and hydrocodone- and oxycodone-acetaminophen combinations) and cases of liver failure in patients taking <4 g/day, the U.S. Food and Drug Administration (FDA) requested in 2011 that manufacturers limit combination products to 325 mg. Soon after, the manufacturer changed the maximum daily dose to 3 g/day, and in 2014 the FDA asked prescribers and pharmacies to discontinue prescribing or dispensing products with more than 325 mg of acetaminophen.

In 2011 the U.S. Food and Drug Administration requested that manufacturers of acetaminophen-containing products limit those products to a maximum of 325 mg of acetaminophen per dose to decrease the chance of hepatic toxicity.
Mechanism of Action
The mechanism of action of acetaminophen is unknown, although there is strong evidence for a central site of action. Multiple sites of action have been postulated, including COX-1, -2, and -3 inhibition, and modulation of the opioidergic, serotonergic, and endocannabinoid systems (2).
Pharmacokinetics and Pharmacodynamics
Acetaminophen is highly bioavailable after oral administration, and it crosses the blood–brain barrier. Time to peak effect is approximately 20 minutes (2).
Metabolism and Excretion
The major metabolic pathway of acetaminophen is glucuronidation and sulfation in the liver. The nontoxic conjugates that are formed are largely excreted in the urine and bile. Between 5% and 9% of acetaminophen is oxidized by the cytochrome P450 (CYP) system, resulting in N-acetyl-p-benzoquinone-imine (NAPQI), the toxic metabolite that can cause hepatocellular injury if the hepatic detoxification process is overwhelmed by a large dose of acetaminophen. NAPQI is highly reactive and is rapidly metabolized by conjugation with glutathione transferase to a nontoxic compound that is eventually excreted as mercapturic acid and cysteine conjugates. Approximately 2% of ingested acetaminophen is excreted unchanged by the kidneys (3).
Drug Interactions and Adverse Effects
Acetaminophen is a dose-related hepatotoxin due to the small amount of NAPQI produced. Depleted glutathione stores due to overdose, malnutrition, or alcohol ingestion may increase the risk of hepatotoxicity. Drugs that induce the CYPs, such as anticonvulsants (phenytoin, carbamazepine, phenobarbital), and isoniazid may increase the risk of acetaminophen toxicity, although the evidence for clinically significant drug–acetaminophen interactions is incomplete. Acetaminophen poisoning was the leading cause of acute liver failure between 1995 and 2003 in the United States, with 48% of acetaminophen-related cases due to unintentional overdose (3). Fifty-one percent of patients had used a single over-the-counter acetaminophen product.
B. Acetylsalicylic Acid
Indications, Contraindications, and Dose
Acetylsalicylic acid (also called aspirin or ASA) can be used for relief from mild to moderate pain. It has both anti-inflammatory and antipyretic effects. Today it is prescribed most commonly for its antiplatelet effects and has indications including acute myocardial infarction, unstable angina, transient ischemic attacks, prevention of arterial and venous thrombosis, and platelet disorders. Aspirin irreversibly inhibits platelets aggregation by inhibiting platelet thromboxane A2 synthesis and by preventing adenosine diphosphate release from platelets. Its antiplatelet effect lasts for the life of the platelet (8 to 10 days) (4). Contraindications to aspirin use include the presence of gastric and duodenal ulcers, as aspirin may cause upper gastrointestinal (GI) bleeding from erosive gastritis. Enteric coating, taking aspirin and other NSAIDs with meals, histamine 2 antagonists, and proton pump inhibitors are helpful in minimizing mucosal damage. Aspirin is contraindicated in children due to the association with Reye syndrome, a life-threatening acute liver failure seen after certain viral infections. The antithrombotic effects of aspirin do not seem to be dose dependent, while the GI side effects and risk of bleeding increase with increasing doses. Therefore, a daily dose of 81 to 325 mg is prescribed for most indications (4).
Mechanism of Action
Aspirin is a nonselective, irreversible COX inhibitor. It also affects the kallikrein system, decreasing granulocyte attachment to injured vessels, among other effects (4).
Pharmacokinetics and Pharmacodynamics
Aspirin can be administered orally or per rectum, with a 40% to 50% bioavailability. Aspirin has a short (15 minutes) plasma half-life, while the plasma half-life of its primary metabolite, salicylic acid, is much longer and dose dependent, between 2 and 12 hours. Of note is the difference between aspirin’s short pharmacokinetic half-life and its prolonged pharmacodynamic effects due to the irreversible platelet inhibition.
Metabolism and Excretion
Aspirin undergoes extensive first-pass metabolism and is metabolized to salicylic acid, which has less pharmacologic activity in the liver, plasma, and red blood cells. Aspirin is highly bound to albumin. Only a small amount of aspirin is excreted unchanged in the urine; the rest is excreted as salicylic acid or glycine.
Drug Interactions and Adverse Effects
Aspirin can erode gastric mucosa, cause acute tubular necrosis secondary to decreased renal blood flow, and precipitate bronchospasm in patients with asthma or aspirin sensitivity. Because aspirin blocks the cyclooxygenase enzymes, substrate is shifted into the alternative pathway, thus increasing the generation of leukotrienes. Aspirin overdose may present with acid-base abnormalities, renal failure, dehydration, abnormal blood glucose, seizures, or coma. These patients require management in the intensive care unit with alkaline diuresis to promote salicylate excretion and may require hemodialysis. Activated charcoal may prevent absorption of aspirin after overdose.
C. Other Nonsteroidal Anti-inflammatories: Ibuprofen, Ketorolac, and Celecoxib
Nonselective NSAIDs (nsNSAIDs) carry a side-effect profile that includes dose-related inhibition of platelet aggregation, GI bleeding, renal dysfunction, altered bone healing, hypertension, and bronchospasm. The COX-2 inhibitors were developed in an attempt to eliminate some of these adverse effects, particularly the GI side effects and interference with platelet aggregation. In 2004, the manufacturer voluntarily withdrew rofecoxib from the market after an interim analysis of a long-term study in patients with colon polyps demonstrated an increased risk for cardiovascular events, including myocardial infarction and stroke. For similar reasons, the FDA asked the manufacturer to voluntarily withdraw valdecoxib from the market in 2005. Celecoxib, with its intermediate levels of COX-2 selectivity compared with rofecoxib and valdecoxib, remains the only COX-2 inhibitor available in the United States. The FDA groups all NSAIDs, whether COX-2 selective NSAIDs or nsNSAIDs, together with regard to skin, cardiovascular, renal, and GI side effects.

The rate of upper gastrointestinal complications is highest with ketorolac compared with other nonsteroidal anti-inflammatories and therefore it should not be used for more than 5 consecutive days.
Indications, Contraindications, and Dose
Ibuprofen, ketorolac, and celecoxib are indicated for pain, inflammation, and fever. When used in the perioperative period, nsNSAIDs demonstrate an opioid-sparing effect. Thus, they decrease the common opioid side effects such as postoperative nausea and vomiting, constipation or ileus, and cardiorespiratory depression (5). Because celecoxib has no effect on platelet function, it is especially useful in the perioperative period. Ketorolac is generally considered to be contraindicated in tonsillectomy, adenoidectomy, total joint replacements, and major plastic surgery because of the increased risk of bleeding on the “raw” surface areas. Note, however, that in the studies demonstrating increased blood loss, ketorolac had been administered pre-emptively, either before surgical incision or before achieving primary hemostasis. No controlled studies in the peer-reviewed literature demonstrate an increase in blood loss when ketorolac was administered near the end of surgery or postoperatively (5). A 2013 meta-analysis published by the Cochrane Collaboration found both an increased and decreased risk of bleeding after pediatric tonsillectomy. It concluded that although there is insufficient evidence to exclude an increased risk of bleeding when NSAIDs are administered during pediatric tonsillectomy, the use of NSAIDs did confer the benefit of a decrease in postoperative vomiting (6). Concerns about bone healing from animal models and human experiments have long limited the use of NSAIDs in orthopedic surgery. However, a meta-analysis revealed no increased risk of nonunion associated with NSAIDs and spinal fusion surgery when only high-quality studies were included (5). Other studies show poor fracture healing when nsNSAIDs as well as COX-2 inhibitors are used. Some experts advocate avoiding NSAID use in patients or procedures with a high risk of nonunion.
The risk of GI complications is present for all NSAIDs, including COX-2 inhibitors. A recent meta-analysis concluded that the relative risk for upper GI complications of celecoxib and ibuprofen are among the lowest of the NSAIDs studied, while ketorolac had one of the highest relative risks for upper GI complications (7). Proton pump inhibitors offer effective protection against peptic ulcers when administered with nsNSAIDs, but COX-2 selective NSAIDs may be more protective of the small intestine.
Ibuprofen and ketorolac are contraindicated in patients with renal insufficiency, as even short courses (<5 days) can produce transient reductions in renal function. This transient reduction is clinically unimportant in patients with normal preoperative renal function, and there are no reports of renal failure when ketorolac was administered for ≤5 days (5). Celecoxib appears to have the same rate of renal dysfunction as the nsNSAIDs. Celecoxib is contraindicated after coronary artery bypass surgery because it is associated with a greater overall risk of adverse events compared with placebo. Celecoxib is also contraindicated in patients with active gastrointestinal bleeding, history of allergy to sulfonamides (due to the presence of a sulfa moiety), and history of asthma or allergic-type reactions to NSAIDs. However, celecoxib has no effect on platelet function or bleeding time.
Ibuprofen is available over the counter with a recommended oral dose of 200 to 400 mg every 6 hours. Inflammatory conditions may require 400 to 800 mg orally every 6 to 8 hours. Children should receive 5 to 10 mg/kg every 6 to 8 hours. In 2009, an intravenous preparation was approved by the FDA. Ketorolac is dosed at 15 to 30 mg intramuscularly or intravenously every 6 hours for no more than 5 days. A single dose of 60 mg may be used. The pediatric dose is 0.5 mg/kg. Celecoxib is available in 50 mg, 100 mg, 200 mg, and 400 mg capsules. Acute pain in adults is dosed at 400 mg orally, then 200 mg orally twice a day. It is approved for use in children older than 2 years old. For children weighing 10 to 25 kg, the dose is 50 mg twice a day; patients weighing >25 kg may receive 100 mg twice a day.
Mechanism of Action
Ibuprofen and ketorolac are nsNSAIDs that inhibit the cyclooxygenase enzymes (see above). Celecoxib is a COX-2 inhibitor, predominantly inhibiting the COX-2 isoform of the cyclooxygenase enzyme.
Pharmacokinetics and Pharmacodynamics
Ibuprofen has close to 100% oral bioavailability and a time to peak effect of 1 to 2 hours. Seventy percent to 90% of the dose is excreted within 24 hours. It is highly protein bound, as are the other conventional NSAIDs. Protein binding may be an important determinant in its distribution into the synovial fluid, thus, its efficacy in rheumatoid arthritis. Ibuprofen distributes into the cerebrospinal fluid and has a longer half-life in the cerebrospinal fluid than in the plasma.
Ketorolac has a rapid onset when administered intravenously, with a time to peak plasma concentrations of 5 minutes. The half-life of ketorolac is approximately 5 hours, and more than 90% is excreted within 2 days (8). Celecoxib reaches peak plasma concentrations in 2 to 4 hours after oral administration and is extensively protein bound.
Metabolism and Excretion
Elimination and metabolism of NSAIDs occur largely through hepatic biotransformation and renal excretion. Patients with hepatic and renal disease may have higher and more prolonged plasma concentrations. Ibuprofen is oxidized by the CYP enzymes, as well as conjugated with glucuronic acid in the liver. The drug and its metabolites are rapidly excreted in both urine and feces. Excretion in breast milk is minimal, and the American Academy of Pediatrics considers ibuprofen to be compatible with breastfeeding. Ketorolac is metabolized by glucuronidation, and more than 90% is excreted in the urine within 2 days. Celecoxib undergoes biotransformation to carbolic acid and glucuronide metabolites, which are excreted in urine and feces. It is metabolized by the CYP enzymes and has an elimination half-life of about 11 hours.
Drug Interactions and Adverse Effects
The nsNSAIDs carry a side-effect profile that includes dose-related inhibition of platelet aggregation, GI bleeding, renal dysfunction, bone healing, hypertension, and bronchospasm. Short-term (<5 days) use of ketorolac does not increase the risk of perioperative complications, except the risk of bleeding in high-risk surgeries with raw surface areas (tonsillectomy or adenoidectomy, major plastic surgery, total joint replacement). Ketorolac demonstrates a high rate of GI ulceration, which limits its use to <5 days.
The cardiovascular risk of COX-2 inhibitors has been a subject of much concern since two studies investigating colon polyp prevention with COX-2 inhibitors were stopped early because of significantly increased risk for the composite of cardiovascular death, myocardial infarction, stroke, or heart failure. Two COX-2 inhibitors (rofecoxib and valdecoxib) were taken off the market in response.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


