What Is the Influence of Hypercoagulability in Thrombosis?
Thrombosis in either the venous or arterial circulation may have catastrophic consequences. In the venous circulation, three vascular conditions occurring singly or, more often, in concert, put patients at increased risk for thrombosis. These include (a) stasis, (b) injury to vascular endothelium, and (c) hypercoagulability. Hypercoagulability may be thought of as a state of exaggerated activation of coagulation. A comparable triad that predisposes to arterial thrombi includes flow limitation from atherosclerosis, plaque rupture as a uniquely arterial form of endothelial injury, and arterial hypercoagulability, an entity just beginning to be understood. Fundamental to the prevention and management of hypercoagulability is a better understanding of thrombosis and its endogenous regulators. Accordingly, this review will begin by describing the most current model for the initiation and propagation of clotting, together with the role of natural anticoagulants, the activity of which is critical to the prevention of thrombosis. Using that model, the etiology of different hypercoagulable states will be explored.
Sources of hypercoagulability can be divided into two major classes: (a) a congenital predisposition caused by one or more genetic abnormalities, often referred to as thrombophilia and (b) acquired, or environmental hypercoagulability.
Genetic mutations that reduce the functional levels of endogenous anticoagulants—antithrombin, protein C, and protein S—have long been known to predispose to venous thromboembolism (VTE). However, new genetic abnormalities are being discovered that contribute to hypercoagulability. Indeed, with appropriate testing, causes of thrombophilia are being detected in as many as 50% of VTE cases. However, genetic factors are only rarely the sole contributor to VTE. Genetic sources of thrombophilia create a lifelong hypercoagulable state that give rise to clinical VTE only episodically. In most cases of VTE, hypercoagulability—acquired or environmental—serves as a triggering factor. As anesthesiologists are often actively involved in clinical events that create acquired hypercoagulability, for example, pregnancy, surgery, and malignancy, it is important to stay abreast of advances in the understanding and treatment of thrombosis.
Historically, blood clotting has been viewed as a series of enzymatic reactions in which the participants, sequence, and regulation were independent of location. Increasingly, recognition of the unique conditions operating in the arterial circulation, including (a) the high velocity of blood flow, creating shear forces in vessels of smaller diameter, (b) its complex rheology at vessel branch points, and (c) its unique anticoagulant requirements (i.e., better efficacy of antiplatelet agents as opposed to antithrombin agents), has mandated the development of an arterial clotting model distinct from that operating in the venous circulation.
1 In concert with this improved understanding of the physiology of arterial hemostasis has come an appreciation for how tightly regulated the controls on this system must be maintained. Indeed, given the spectrum of clinical complications associated with defects in arterial hemostasis, including catastrophic blood loss at one end and infarction at the other, it is not surprising that multiple interdependent factors keep a very tight rein on this process. Accordingly, although there are conditions that give rise to both venous and arterial hypercoagulability, a subset of heritable and acquired conditions exist that
uniquely predispose to arterial thrombosis. The current understanding in the field of arterial hypercoagulability is not as advanced as that of venous hypercoagulability, but has the potential to significantly improve the medical treatment of heart disease and stroke.
How Is Venous Thromboembolism Defined and Assessed?
VTE has an annual incidence that exceeds 1 per 1,000, with approximately 2 million cases in the United States alone, and more than 150,000 deaths per year resulting from pulmonary embolism. These statistics put VTE in a class with clinical events such as stroke. As early as the late 1800s, Virchow identified the three vascular conditions predisposing to VTE: (a) stasis, (b) injury to vascular endothelium, and (c) hypercoagulability. The first two made intuitive sense, but very little was known at the time about the pathophysiology of hypercoagulability. A better understanding of the physiology of normal hemostasis, especially factors that regulate the velocity of clot growth and its composition, has paved the way for recognizing the genetic and acquired conditions that create a state of heightened coagulation activation, that is, hypercoagulability. Accordingly, this review will begin with an overview of the current model of normal coagulation.
How Does Normal Venous Coagulation Occur? The Two-Phase Model
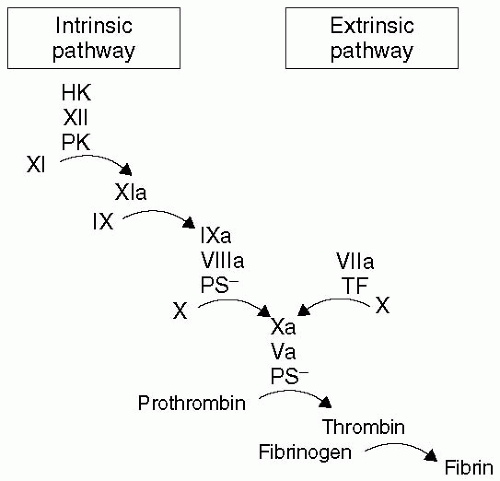
Fifty years ago, two groups simultaneously described the “waterfall” or “cascade” model (see
Fig. 38.1) of soluble coagulation.
2,
3 This model allowed great strides to be made in identifying the series of proteolytic reactions that culminate in a fibrin clot. The cascade model seemed to fit with the clotting assays that were developed to guide warfarin and heparin dosing. These tests became the gold standard for measuring soluble coagulation. Although this cascade model is practical in clinical scenarios associated with deficits of one or more factors, it fails to explain the bleeding diathesis associated with hemophilia, and offers little insight into thrombophilias. As will be emphasized later in the chapter, the critical determinants of whether a clot will form—be it an appropriate response to bleeding or a pathologic event—are the efficiency and velocity of each of the reaction steps (e.g., how much, how fast).
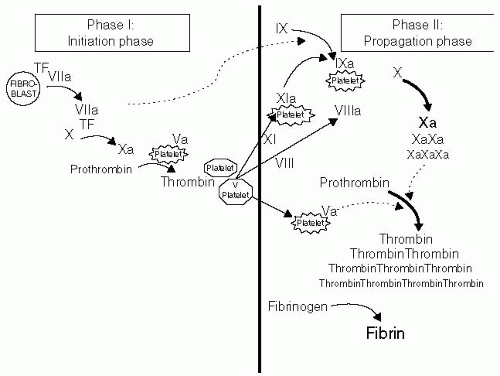
Recent advances in cell-based research models have made significant strides in clarifying the dynamics of coagulation.
4 In vivo coagulation follows exposure of the blood to a source of tissue factor (TF), typically on the surface of a fibroblast or other subendothelial cell in contact with blood elements through damage to the endothelial cells lining the vessel lumen. The intrinsic, or contact, pathway of coagulation plays no role in these earliest clotting events. Tissue factor-initiated coagulation has two phases: An
initiation phase and a
propagation phase5,
6 (see
Fig. 38.2).
The initiation phase begins as exposed TF binds to factor VIIa, picomolar amounts of which are present in the circulation at all times. This VIIa-TF complex catalyzes the conversion of very small amounts of factor X to Xa, which in turn generate nanomolar amounts of thrombin. Collagen exposed in the subendothelium is capable of activating nearby circulating platelets, causing them to expose phosphatidylserine and release factor V, thereby increasing prothrombinase formation and thus the likelihood of thrombin generation.
The seemingly trivial amount of thrombin formed during initiation sparks the inception of the propagation phase, the successful completion of which culminates in explosive thrombin generation and, ultimately, fibrin deposition. More than 96% of the total thrombin generated during clotting occurs during propagation. The commonly used laboratory tests of soluble coagulation only measure the kinetics of the initiation phase.
5 The prothrombin time (PT) and activated partial thromboplastin time (aPTT) both have as their endpoints the first appearance of fibrin gel, which occurs with <5% of the total reaction complete. Therefore, the fibrin clotting that signals completion of the PT/aPTT occurs when only minimal thrombin levels have been formed. These tests are sensitive at detecting complete deficiencies in clotting factors (e.g., hemophilia) and guiding warfarin/heparin
therapy; however, they do not model the entire sequence of events necessary for effective hemostasis. They fail to give information relevant to thrombin generation during the propagation phase, which determines whether a persistent clot will form, or whether endogenous anticoagulants and fibrinolytic regulators are able to constrain excess clot growth.
Thrombin generated during the initiation phase is a potent platelet activator, thereby providing both an activated platelet surface membrane and platelet-released factor V (which thrombin promptly converts to Va). Factor VIII, conveniently brought to the bleeding site by its carrier, the von Willebrand factor (vWF), is also activated by thrombin—a step that causes its release by vWF. FVIIIa then complexes with the picomolar amounts of factor IXa, also generated by the TF-VIIa complex during the initiation phase, forming the FVIIIa-IXa complex, a pivotal point in the successful generation of a clot.
The formation of the FVIIIa-IXa complex on the platelet surface heralds the switch from FXa generation by the TF-VIIa complex to the intrinsic Xase pathway. This switch is of enormous kinetic advantage, with the intrinsic Xase complex exhibiting approximately a 50-fold greater efficiency at Xa generation. The bleeding diathesis associated with hemophilia is testament to the hemostatic importance of the exuberant thrombin generation formed during the propagation phase.
7 Although congenital deficiencies in VIII and IX do prolong the aPTT, it is the thrombin generation in the propagation phase—a function not evaluated by the aPTT—that is most impaired in hemophilia.
The activated platelet, stimulated by thrombin formed in the initiation phase, expresses membrane receptors for VIIIa and IXa. When these active proteases are bound in combination with negatively charged phosphatidylserine, the resulting enzyme complex enhances the binding of its substrate, factor X. The speed of X activation by this complex is also increased by platelet binding; indeed, when compared to the reaction speed of free proteases in solution, assembly of the entire reaction on the platelet membrane increases the catalytic efficiency of X activation by approximately 13 million-fold.
Assembly of the prothrombinase complex is similarly dependent on the activated platelet surface for optimum activity. Much of factor V released by the activated platelet (subsequently thrombin-activated) stays bound to the platelet surface. Like the Xase complex, the platelet-bound prothrombinase complex (i.e., Xa-Va-phosphatidylserine) enhances prothrombin activation to thrombin 300,000-fold faster than free Xa and Va acting on prothrombin formed in solution. Platelet-bound Xa is the rate-limiting reagent in prothrombin cleavage. The substrate for this enzyme complex, prothrombin, binds to GPIIb/IIIa on both activated and inactivated platelets, potentially providing a source for thrombin generation in both the initiation and propagation phases.
Evidence that factor XI further amplifies the propagation phase is growing.
7 As noted in the preceding text, Xa levels are rate limiting to the prothrombinase complex, particularly once the switch is made from extrinsic (FVIIa:TF complex) to intrinsic (FVIIIa:FIXa complex) Xase. Small amounts of IXa can be generated by the TF-VIIa complex, but its ability to sustain IXa generation is limited by its endogenous inhibitor, tissue factor pathway inhibitor (TFPI).
7 To generate Xa in amounts sufficient to fuel the propagation phase, an alternative and kinetically superior source of IXa is needed. Factor XI is another zymogen activated by the minute amounts of thrombin generated during initiation, but this activation only occurs on the activated platelet surface.
7 Platelet-bound FXIa is ideally located to activate FIX, which also binds to the platelet surface, helping to speed and localize this activity. Additionally, binding to the platelet surface protects FXIa from its inhibitor, protease nexin 2. Therefore, FXIa generation on the activated platelet is key to providing sufficient FIXa to maintain Xa generation through the catalytically more efficient intrinsic Xase complex.
In the venous circulation, the kinetic advantage of coagulation cascade assembly on the activated platelet surface is readily apparent; recent
in vitro work has clarified the minimum platelet count (i.e., the
dose of platelets) necessary for the reaction sequence to proceed.
8 When all coagulation zymogens are present, thrombin is not generated unless platelets are present as a source of phospholipid. Once platelets are added, thrombin generation begins and grows with increasing numbers
of platelets, up to a threshold of 10,000 platelets per µL. Increases in platelets beyond this number have no effect on the efficiency of the reaction, suggesting that, as had been empirically observed in thrombocytopenic patients, the platelet level must decrease to very low levels (i.e., <10,000 per µL) to increase the risk of venous bleeding. This contrasts sharply with the arterial circulation where the minimum platelet count needed to ensure hemostasis for operative procedures is at least five times that number.
What Are Endogenous Anticoagulants?
In addition to providing a kinetically favorable orientation, the platelet surface membrane protects the active coagulation enzymes from inactivation by proteases circulating in plasma. During the initiation phase, binding of FXa to the platelet membrane protects it from inactivation by both TFPI and antithrombin. Normal plasma levels of both TFPI and antithrombin inhibit soluble FXa so efficiently that its plasma half-life is ≤1 minute.
4 Preservation of small amounts of FXa that are generated during this “make or break” stage of coagulation is critical to formation of the nanomolar amounts of thrombin needed to begin the propagation phase. Similarly, the thrombin formed during the initiation phase must also be protected from inactivation by antithrombin. Antithrombin is present at over twice the concentration (3.2 µmol per L) of the highest concentration of thrombin reached during the propagation phase (1.4 µmol per L) and approximately 1,000 times the nanomolar amounts of thrombin generated during the initiation phase. Without the protection conferred by the platelet membrane, normal plasma levels of antithrombin inhibit soluble thrombin, with a half-life of <1 minute. Therefore, thrombin generated during the initiation phase is critically dependent on protection by the activated platelet membrane to have sufficient time to transition from the initiation phase to the propagation phase.
The other critical physiologic regulator of thrombin generation is protein C. Unlike antithrombin, which circulates in an active form (albeit capable of enhanced kinetics when bound to heparin or endogenous heparan sulfate), protein C requires activation by thrombinstimulated endothelial cell thrombomodulin. Activated protein C (APC) then acts in concert with its cofactor, protein S, to limit the rate of thrombin generation by inactivating the essential procoagulant cofactors, FVa and FVIIIa.
9 Again, the activated platelet membrane promotes clot success by protecting FVIIIa and FVa from inactivation by APC.
10