Chapter 23 Non–Laryngeal Mask Airway Supraglottic Airway Devices
I Introduction
Nothing is more fundamental to the practice of general anesthesia than the maintenance of a clear upper airway. The choice of device depends on several factors, including access to the airway, duration of surgery, and risk factors for aspiration. After placement, the cuffed ETT provides a secure airway and protects against aspiration, but placement and removal of an ETT require training and judgment. Although ETTs typically are used without incident, complications ranging from trivial to life-threatening can occur.1
Advanced airway management depends on many airway devices, several of which have been included in the American Society of Anesthesiologists (ASA) difficult airway algorithm.2 The Classic laryngeal mask airway (LMA Classic, LMA North America, San Diego, CA) was introduced into clinical practice in 1988. Since then and particularly in the past 10 years, there has been an explosion of supraglottic airway devices (SADs) designed to compete with the LMA Classic, especially single-use devices. The introduction of single-use devices has been driven by concern about the sterility of cleaned, reusable devices (e.g., elimination of proteinaceous material, risk of transmission of prion disease) and the inability to recycle the device enough to be cost-effective. More than 20 manufacturers produce single-use LMs. Other designs of SADs have been introduced, and they are the main focus of this chapter.
II Nomenclature
The term supraglottic airway device (SAD) is used to describe a group of airway devices designed to establish and maintain a clear airway during anesthesia. SADs have several roles, including maintenance of the airway during spontaneously breathing or controlled-ventilation anesthesia, airway rescue after failed intubation or out of the hospital, use during cardiopulmonary resuscitation, and use as a conduit to assist difficult tracheal intubation. Brimacombe recommended that the term extraglottic airway be used, because many of these devices have components that are infraglottic (i.e., hypopharynx and upper esophagus).3 This textbook describes all airway devices that have a ventilation orifice or orifices above the glottis as supraglottic and those that deliver anesthetic gases or oxygen below the vocal cords (e.g., transtracheal jet ventilation, cricothyrotomy) as infraglottic. Other terms and acronyms include supraglottic airway (SGA), extraglottic airway device (EAD), and periglottic airway device (PAD), but SAD is more widely accepted and is used in this chapter.
Brimacombe and Miller suggested there should be a classification system for this increasingly complex family of devices. Miller4 described three main sealing mechanisms: cuffed perilaryngeal sealers, cuffed pharyngeal sealers, and cuffless, anatomically preshaped sealers. Further subdivision can be made by considering whether the device is single use or reusable and whether protection from aspiration of gastric contents is offered. The practical value of this type of classification is uncertain. Chapters 22 and 27 review the LMA, its variants, and the Combitube.
Second-generation SADs have been designed with safety in mind, and they incorporate design features that aim to reduce the risk of aspiration.5 They include the ProSeal LMA (PLMA), i-gel, LMA Supreme, Laryngeal Tube Suction II (LTS-II), disposable version of the LTS (LTS-D), the Streamlined Liner of the Pharynx Airway (SLIPA), and the Baska mask. The efficacy of several of these designs has not been proven.
III Limitations of the Classic Laryngeal Mask Airway
Prior to 1988, choices of airway devices essentially were limited to the face mask and endotracheal tube (ETT). The LMA Classic was designed by Archie Brain in the United Kingdom in the early 1980s, and it was introduced into anesthetic practice in 1988. Its introduction revolutionized airway management (Fig. 23-1). It was soon recognized to be a suitable device to use for many cases that previously were managed with a face mask or an ETT, because the LMA Classic had many advantages over both devices.6 It has been used in approximately 200 million episodes of anesthesia globally. More than 2500 studies on the device have been published. The LMA Classic is considered the benchmark against which other SADs are judged. A 2008-2009 UK census found that 56% of all episodes of general anesthesia were delivered with a SAD as the primary airway,7 and 90% of the devices were LMAs and LMs.
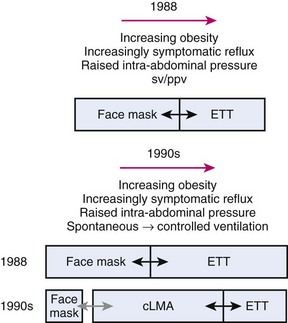
A Problems with Controlled Ventilation
The LMA Classic usually seals the pharynx with a pressure of 16 to 24 cm H2O, and this airway leak pressure is rarely above 30 cm H2O. This relatively low- pressure seal means that when positive pressure is applied to the LMA Classic, gas leakage is common. Studies have shown a 5% failure rate for achieving an expired tidal volume of 10 mL/kg,8 an audible leak in 48% of patients when ventilating to peak pressures of 17 to 19 cm H2O, and a detectable leak rate as high as 90%, with an 8% failure rate for adequate ventilation.9,10 Devitt and colleagues applied increasing peak airway pressures while ventilating through an LMA Classic. They found that as the airway pressure rose from 15 to 30 cm H2O, the incidence of audible leak rose from 25% to 95%, and the leak fraction ([inspired minute volume−expired minute volume]/inspired minute volume) rose from 13% to 27%.11 As the airway pressure increased, the incidence of airway leak into patients’ stomachs rose from 2% to 35%. These findings indicate that the LMA Classic has a relatively low-pressure airway (pharyngeal) seal and that as higher airway pressures are applied, there is a risk of loss of ventilating gases and gastric inflation. Loss of ventilating gases is associated with hypoventilation, loss of anesthetic agent, and environmental pollution, and gastric inflation that may increase the risk of regurgitation.
B Problems with Airway Protection
The LMA Classic is not regarded as providing protection against aspiration of regurgitated gastric contents and is contraindicated for patients who are not fasted or who may have a full stomach. The LMA Classic has a pharyngeal seal that is usually in the range of 16 to 24 cm H2O. Its tip obturates the upper esophagus, and it has an esophageal seal of 40 to 50 cm H2O, but it has no drain tube.12 Despite this design, cadaver work shows the LMA Classic protects the glottis from regurgitant esophageal fluid considerably more efficiently than the unprotected airway.13
Soon after the introduction of the LMA Classic, several small studies raised concerns about the ability of the LMA Classic to protect the airway from regurgitant matter and therefore from pulmonary aspiration. Early concerns were raised that the LMA Classic, sitting at the back of the throat, might stimulate a swallowing reflex, especially during light planes of anesthesia, leading to relaxation of the upper and lower esophageal sphincters and increasing the risk of regurgitation and aspiration. A physiologic study recorded a fall in the lower esophageal barrier pressure of 4 cm H2O during LMA Classic anesthesia, compared with a 2 cm H2O rise during face mask anesthesia.14 A study using swallowed methylene blue capsules demonstrated a 25% incidence of soiling of the inner portion of the LMA Classic on removal, and a small study reported a 2% incidence of aspiration, which occurred during spontaneous and controlled ventilation.15,16
As experience has accumulated, the evidence of a fundamental problem with aspiration has been reevaluated. Until 2004, 16 years after its introduction, there were no published reports of fatal aspiration during use of an LMA Classic. In 2004, Keller and colleagues published a series of three cases of serious morbidity, including one death from aspiration during LMA Classic anesthesia.17 Each case had risk factors for aspiration, and on reviewing all 20 published reports of aspiration during use of an LMA Classic, the investigators found identifiable risk factors in 19 of 20 cases. In the accompanying editorial, Asai listed more than 40 factors that increased the risk of aspiration.18 Several large studies have shown a low rate of aspiration; Verghese and Brimacombe reported a series of 11,910 uses (40% with controlled ventilation, 19% during intra-abdominal surgery, and 5% with a duration longer than 2 hours).19 Insertion success rate was 99.8%, the incidence of airway-related critical incidents was 0.16% during spontaneous ventilation and 0.14% during controlled ventilation, and there was one case of aspiration. Bernardini and Natalini reported three aspirations in a series of 35,630 LMA Classic uses for controlled ventilation (1 of 11,877).20 In an editorial, Sidaras and Hunter21 estimated an incidence of confirmed pulmonary aspiration during LMA Classic use of 1 in 11,000, and Brimacombe and Berry’s meta-analysis calculated a risk during elective surgery of 1 case in 4300 operations.6 This is similar to the rate of aspiration reported by Warner and colleagues in a study of 214,000 patients predating use of the LMA, in which aspiration occurred in 1 in 4000 elective operations.22
Although the risk of aspiration is relatively low in expert hands, this rate is achieved primarily by careful and appropriate case selection, expert insertion, and meticulous management of the airway after insertion. The Fourth National Audit Project of the Royal College of Anaesthetists and Difficult Airway Society (NAP4) in the United Kingdom studied major airway complications of 2.9 million episodes of general anesthesia and found that aspiration was the most common cause of airway-related deaths.1 One third of these complications occurred during maintenance with a LM or LMA in place, and for many of the patients, the risk of aspiration made this unwise.
C Problems with Accessing the Airway for Intubation
The LMA Classic sits over the vocal cords in more than 90% of cases and may be used as a conduit for intubation, but several factors limit the ease of this application.23 The internal lumen of the device is relatively narrow, limiting the size of ETT that can be passed. Size 4 and 5 LMA Classic devices accommodate most manufacturers’ cuffed ETTs with internal diameters (IDs) of 6.0 and 6.5 mm, respectively. A tube of adequate length must be used to exit the LMA Classic and reach the midtrachea; an ETT of approximately 29 cm can be placed through a size 5 LMA Classic. Across the distal end of the airway tube are two flexible bars forming a grill that prevents the tongue from impeding insertion and the epiglottis from causing obstruction after placement; these bars may act as an impediment to intubation through an LMA Classic. The angle at which an ETT exits the mask of the LMA Classic means that blind insertion frequently leads to esophageal intubation. Brimacombe reported blind intubation with an ETT through the LMA Classic to have a first-time success rate of 52% and overall success rate of 59%.24 Use of a bougie is less successful (32% of first attempts and 45% overall), and even fiberoptically guided techniques have a failure rate of 18%. After intubation has been achieved, removal of the LMA Classic without displacement of the ETT is cumbersome. Overall, direct intubation through the LMA Classic is a far from ideal technique.
The technique is dramatically improved if an Aintree intubation catheter (AIC, Cook Critical Care, Bloomington, IN) is used.25 The hollow AIC (ID of 4.6 mm, external diameter [ED] of 7.0 mm, length of 46 cm) is placed over a fiberscope, the scope and AIC are negotiated through the LMA Classic into the midtrachea, and the fiberscope then is removed, followed by removal of the LMA Classic. Care must be taken to ensure the AIC is not advanced too far, especially if gases are passed through it, as this risks barotraumas. This technique can be performed with or without the use of a Bodai adapter (Sontek Medical, Lexington, MA). The Bodai adapter allows oxygen and gas administration through the attached breathing circuit during exchange of the LMA to an ETT. The AIC remains in place, and a suitably sized lubricated ETT is then advanced over the catheter. Although the AIC technique does not appear in current airway guidelines, its use is simple, has a high success rate, and is widely reported.26,27
D Reusable Design
The LMA Classic is reusable and designed to be used up to 40 times. An in vitro study suggested that the LMA Classic and ProSeal LMA may be reused up to an average of 130 and 80 times, respectively, before showing signs of failing the preuse tests recommended by the manufacturer, and in vivo work supports use up to 60 times.28,29
After use, the LMA Classic is cleaned (decontaminated) before sterilization by autoclave (up to 137° C for 3 minutes with the cuff fully deflated), and it is stored in sterile packaging thereafter. A 2001 bench-top study demonstrated that routine decontamination and sterilization failed to remove all proteinaceous material from airway devices and from the LMA Classic in particular.30 At the same time, there was increasing public awareness about variant Creutzfeldt-Jakob disease (vCJD), especially in the United Kingdom. Concerns grew that residual prions, the infective, misfolded proteins responsible for vCJD, might remain and be passed from patient to patient. Several national bodies recommended using single-use devices “wherever possible,”31 even though the estimated risk of such cross-contamination was 1 to 10 cases in 100,000 patients.32 Brimacombe and coworkers described the rush toward single-use LMs as “driven by fears of the unknown and scientific misinformation.”33 Since then, the risk of vCJD has fallen dramatically, and the risk of transmission is likely to be vanishingly small.34 This risk must be balanced against other risks introduced by alternative equipment.35 Blunt and Burchett found that even a small deterioration in safety as a result of using a single-use device of poorer quality in place of a reusable device increased the overall risk to patients and went against the recommendation of the Spongiform Encephalopathy Advisory Committee (SEAC).32
E Absence of a Bite Block
The LMA Classic lacks a bite block and is prone to obstruction by biting in the agitated patient during emergence. Use of a bite block (e.g., rolled gauze placed between the molar teeth) is recommended until the LMA Classic is removed, and failure to adhere to this recommendation can lead to airway obstruction, hypoxia, and postobstructive pulmonary edema.36 The LMA Flexible also has no bite block, but the intubating LMA (ILMA), ProSeal, and Supreme LMA do.
IV Efficacy, Safety, and Evaluation of Supraglottic Airway Devices
A small survey of SAD manufacturers in 2003 examined several devices introduced around that time35 and found that the number of patients in whom the device had been used before marketing was less than 150 all in cases but one. In most cases, no trials were published in peer-reviewed journals before launching the product. One device launched in 2001 and remained without published data 18 months later. Only two of seven devices were compared with the LMA Classic in randomized, controlled trials before marketing, and the largest of them enrolled only 60 patients. This situation has not changed, and many later devices have been introduced with little or no trial evidence of their efficacy.
What regulations govern the introduction of new medical devices, particularly airway devices? In the European Union, the use of medical devices is controlled by three European Directives as part of European law.36 Some directives are specifically applicable to airway devices, and adherence is overseen by a regulatory body in each member country. The statutory body has responsibility for ensuring that medical devices do not threaten patients’ health and safety. Statutory requirements are largely harmonized throughout Europe, and compliance with one country’s requirements allows distribution and marketing of a device throughout the European Union. Although many countries have mechanisms that are designed to critically examine the efficacy of new technologies (e.g., National Institute of Clinical Excellence [NICE] in the United Kingdom), these bodies often have specific conditions (e.g., for NICE, new technology for new procedures) such that new (airway) equipment designed to do an old job tends to fall outside their areas of inspection and regulation.
After a device is marketed, clinical trials are not required to demonstrate efficacy or quality of performance. Manufacturers are legally bound to report serious or potentially serious adverse incidents.37 The statutory body requires reporting of incidences in which “malfunction of or deterioration in the characteristics and performance of a device” leads to “actual or potential patient harm.”37 There is also a mechanism for voluntary reporting of incidents by users. Whether these mechanisms lead to reliable reporting of such incidents and whether these schemes identify devices that are poorly designed or underperform is not clear. Formal assessment of performance may come from postmarketing cohort or comparative studies. However, these studies are uncommon, and they usually are published at some interval after a device has been marketed.
A Desirable Features of Supraglottic Airway Devices
Many assume that reusable devices may be replaced by cheaper, single-use devices, and some think that single-use devices are intrinsically preferable. However, many single-use devices differ from the reusable devices they seek to replace in design and in the materials used. Some modifications appear to be minor, but the implications for performance have generally not been evaluated. The work on single-use laryngoscopes and intubation bougies provides evidence that changes in product material may alter performance considerably.38,39 Data on the current versions of the single-use LM and comparisons between these and the LMA Classic remain largely unavailable.
B Efficacy Versus Safety
Safety encompasses avoidance of complications occurring at all stages of anesthesia and afterward. Prevention of aspiration requires a good-quality seal within the laryngopharynx and esophagus (i.e., esophageal seal) to prevent gas leaking into the esophagus and stomach and to prevent regurgitant matter passing from the esophagus into the airway. A functioning drain tube enables regurgitant matter to bypass the larynx and be vented outside, protecting the airway and giving an early indication of regurgitation to the anesthesiologist. Studies have shown that the extent of esophageal seal varies considerably among SADs. Those with a drain tube can effectively vent regurgitant fluid if the drain is not occluded.12,40,41
C Structured Approaches to Evaluation of New Devices
New airway devices should undergo mandatory assessment of manufacturing quality and clinical performance before marketing. The characteristics of the ideal SAD outlined earlier provide a checklist against which function can be assessed. Several methods have been recommended.35,42,43 Cook described a three-stage evaluation process35:
Stage 1. Bench evaluation using manikins or models designed to test function and basic safety
Stage 2. A rigorous cohort study to determine whether the device is effective and to further exclude major concerns about safety
Stage 3. A randomized, controlled trial against the current gold standard for the procedure for which the new device is expected to be used (e.g., LMA Classic, PLMA, ILMA)
In stage 1, the bench models include airway manikins and others, such as those specifically designed to test aspiration risk.44 This stage is limited by lack of fidelity of available manikins.45 With the increasing use of SADs during resuscitation, during out-of-hospital rescue, and by non-anesthesiologists, there is an urgent need to develop realistic manikins for testing and training. Data acquired from such studies require intelligent interpretation and knowledge of the relative performance of different manikins.46–48 Results of manikins studies are considerably limited, and at best, they may be used to evaluate basic information on device performance and durability and to identify major conceptual or design problems. Appropriate bench testing may lead to further development of a device before starting clinical studies.
In stage 2, a cohort study may be used for the first assessment of clinical performance in patients. This approach enables full clinical evaluation of the new device under routine clinical conditions. Functions that can be tested include ease of insertion, pharyngeal seal, airway resistance, stability of the device in different head and neck positions, ease of passage of a gastric tube, positioning of the airway over the larynx, and suitability for fiberscopic or catheter exchange techniques. Learning curves can be examined. A cohort study also enables assessment of function during spontaneous and controlled ventilation and determination of airway trauma or pharyngolaryngeal morbidity. The cohort must be large enough to enable identification of common problems, but unless it is very large, it cannot detect uncommon or rare problems. For instance, for an event that does not occur in a cohort study of n cases, the 95% confidence interval (CI) for frequency of that event is approximately 1 in  .49 For example, if no nerve injuries occur in a cohort study of 100 cases, the upper limit of the 95% CI for risk of nerve injury is 1 in 33. A cohort of at least 100 patients is a reasonable compromise between being large enough to identify important uncommon events and remaining a practical size.
.49 For example, if no nerve injuries occur in a cohort study of 100 cases, the upper limit of the 95% CI for risk of nerve injury is 1 in 33. A cohort of at least 100 patients is a reasonable compromise between being large enough to identify important uncommon events and remaining a practical size.
Stage 3 employs a randomized, controlled trial. After successful completion of bench and cohort evaluations, the need for further modifications of the device should be considered. Significant modifications necessitate repetition of the early evaluations. On successful completion of the early evaluations, the new device should be compared with its best existing competitor. In many cases, this is the LMA Classic. The randomized, controlled trial must be of adequate size to identify clinically important differences in function. Studies may be designed to test the hypothesis that the devices perform differently (i.e., superiority-inferiority trials) or that the test device does not perform significantly less well than the benchmark device (i.e., noninferiority trials).50 Power calculations can be based on data acquired from phase 2, but trials of at least 100 patients provide more comprehensive and clinically useful comparisons. Economic evaluation of cost-effectiveness of the new device may take place at this stage. Data from the three phases of evaluation can be used to determine what role the new airway device has in the market.
A second structured approach to evaluating and choosing new devices was made by Wilkes and colleagues.43 In this proposal, a central body of experts would coordinate research to evaluate new devices, review available evidence and provide national recommendations on devices reaching standards of acceptability. Although potentially of value for a large population (e.g., a country), the barrier it may create to free trade and the likelihood of legal challenges are problems.
The UK Difficult Airway Society (DAS) has proposed a guideline whereby purchasers could adopt a minimum level of evidence before making a pragmatic decision about the purchase or use of an airway device.42 This minimum level of evidence (i.e., level 3b: a case- or historical-controlled cohort study) would form the basis of a professional standard to guide those with responsibility for selecting airway devices.51 Devices without this minimum level of evidence would not be purchased. The investigators argue that widespread adoption of this professional standard would lead to situations in which it was in the interests of manufacturers and purchasers to acquire such evidence and the DAS would support both parties in setting up research with this aim. The strength of this approach lies in purchasers driving the need to raise the evidence bar and manufacturers being encouraged to perform clinical trials at an early stage in device development. This approach is not anticompetitive because it creates no barriers to manufacturers bringing a device to market, but it does raise the level of expectation of the community of purchasers about what they wish to purchase.
Related posts:
 Prehospital Airway Management
Prehospital Airway Management
 Medical-Legal Considerations: The ASA Closed Claims Project
Medical-Legal Considerations: The ASA Closed Claims Project
 Performance of Rigid Bronchoscopy
Performance of Rigid Bronchoscopy
 Nonintubation Management of the Airway: Airway Maneuvers and Mask Ventilation
Nonintubation Management of the Airway: Airway Maneuvers and Mask Ventilation
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


