Evaluating cancer risk within a family is a dynamic process. Individuals may move to a higher-risk group as more cancers develop within the same family over time or as more information is gathered for existing relatives.
In the high-risk group of Lynch and polyposis syndromes, there is a one in two chances of inheriting a lifetime risk of bowel cancer of more than 50 %. The polyposis syndromes can usually be diagnosed without too much difficulty through a recognisable phenotype, but Lynch syndrome can be more difficult because there is no distinguishing feature, only the presence of cancer.
10.3 Lynch Syndrome
This autosomal dominantly inherited condition accounts for approximately 2–4 % of all CRCs and the majority of inherited colonic tumours. Lynch syndrome was previously termed hereditary non-polyposis colorectal cancer (HNPCC), but as some adenomatous polyps can arise in this condition, this term has been largely discarded (Jass 2006).
Lynch syndrome typically presents with CRC in the fourth decade of life with a tendency towards right-sided, poorly differentiated, mucinous colonic tumours with lymphocytic infiltrates. There are often multiple cancers within the colon and elsewhere including the endometrium, stomach, ovaries, renal tract, pancreas, small bowel and brain (Aarnio et al. 1999).
10.3.1 Genetics
Lynch syndrome results from germline mutations in tumour suppressing mismatch repair (MMR) genes coding for proteins that either correct base pair mistakes during DNA replication or stimulate apoptosis when it is irreparable (Jass 2006). Affected individuals inherit a defective copy. MMR function is lost in a cell when the remaining normal gene becomes mutated. A lack of MMR proteins stimulates tumourigenesis by allowing rapid accumulation of mutations in other genes. Microsatellites, regions of short DNA sequence repeats, characteristically become mutated as a result of deficiency in MMR proteins, resulting in microsatellite instability (MSI). MSI is a key feature of tumours that lack MMR and is seen in approximately 15 % of sporadic CRCs as well as those arising in Lynch syndrome.
10.3.2 Diagnosis
A diagnosis of Lynch syndrome can be made using a combination of family history, tumour analysis (to detect MSI) and/or genetic testing. Whilst families fulfilling the Amsterdam II criteria (Box 10.1) may have Lynch syndrome, these will only identify half of those with the syndrome, as 50 % of those affected will fail to meet the criteria. Of those meeting the criteria, half will not have Lynch syndrome (Simmang et al. 1999). Nearly all Lynch syndrome tumours will be MSI high due to MMR mutation and show lack of MMR protein expression on immunohistochemistry (MMR-IHC). The Bethesda criteria (Box 10.2) help in deciding when to test tumours for MSI or MMR-IHC. This will usually identify up to 90 % of people with CRC due to Lynch syndrome (Umar et al. 2004).
Box 10.1. Amsterdam Criteria II (Vasen et al. 1999)
At least three relatives with a Lynch syndrome-associated cancer (colorectal, endometrial, small bowel, ureteral, renal pelvis). One should be a first-degree relative of the other two |
At least two successive generations should be affected |
At least 1 CRC should be diagnosed <50 years |
FAP should be excluded |
Tumours should be verified by pathological examination |
Box 10.2. Bethesda Criteria
CRC diagnosed in a patient <50 years old |
The presence of multiple colorectal or other Lynch syndrome-associated tumours, either synchronous or metachronous at any age |
CRC with high MSI histology in a patient <60 years old |
CRC diagnosed in one or more first-degree relatives with a Lynch syndrome-associated tumour, with one of the cancers diagnosed <50 years of age |
CRC diagnosed in two or more first-degree or second-degree relatives with Lynch syndrome-associated tumours, at any age |
MMR gene mutation detection is expensive, and the decision to test will depend upon individual circumstances. This requires a multidisciplinary team approach with specialist nurses and doctors working within the field of inherited bowel cancer and genetics (Scholefield et al. 1998; Burke et al. 1997). A histopathologist, specialising in inherited CRC, is important when determining which tumours require further testing to achieve an accurate diagnosis. Patients require appropriate counselling, informing them of the possible outcomes from genetic testing, from insurance to family and work life issues. If a mutation is found, appropriate surveillance can be arranged, and other family members can be offered predictive testing to establish whether they carry the abnormal gene. If they do not, they can be discharged from follow-up. In high-risk families where no mutation can be identified, individuals should continue with colonoscopic surveillance.
The complex nature of inherited CRC requires a multidisciplinary approach, particularly in cases of diagnostic difficulty. Regular meetings to discuss complicated cases will require contributions from specialist nurses/counsellors, gastroenterologists/endoscopists, paediatric services, geneticists, radiologists, histopathologists and surgeons. This creates a forum where team members can discuss cases amongst other specialists and consensus can be reached regarding diagnosis, appropriate investigations and management, supported where possible with evidence-based practice. A chairperson directs the team. Where further endoscopies may be required for tissue diagnosis/genetic testing, they can be arranged efficiently and results discussed within the same forum when available. Recommendations can then be communicated to the patient through specialist nurses, gastroenterologists or surgeons as appropriate. This ensures the delivery of a high-quality service where accurate diagnoses can be reached, appropriate surveillance can be arranged and optimal management instituted.
10.3.3 Surveillance
Where screening has been undertaken, significant benefits to life expectancy have been demonstrated in MMR mutation carriers. This is estimated to be an additional 13.5 years, and after prophylactic proctocolectomy, 15.6 years, when compared with no intervention (Syngal et al. 1998).
Colonoscopy is recommended for at-risk individuals every 1–2 years from 25 years of age or 5 years younger than the youngest affected family member, whichever is earlier. This continues until 75 years of age, or where a causative mutation found in a family is excluded from the individual (Vasen et al. 2007). There is no evidence to support surveillance for extracolonic cancers.
10.3.4 Medical Intervention
The Colorectal Adenoma/Carcinoma Prevention Programme 2 (CAPP2) study evaluated aspirin as a chemopreventative agent in Lynch syndrome and found a reduction in CRC rate. However, more data are required to determine optimal dose and treatment duration (Burn et al. 2011).
Some cytotoxic chemotherapy agents are of questionable benefit in Lynch syndrome-associated cancer. It is imperative that oncologists are aware of the underlying diagnosis (Vasen et al. 2007).
10.3.5 Surgical Intervention
Prophylactic surgery should be discussed with high-risk individuals. This may include prophylactic colectomy, hysterectomy and/or bilateral salpingo-oophorectomy. The options for addressing CRC risk include subtotal colectomy or total colectomy with restorative proctocolectomy (RPC).
There is a 16 % risk of metachronous bowel tumours after 10 years follow-up (Ruschoff et al. 1998). For those with a colonic tumour, the surgical options are segmental colonic resection or colectomy with ileorectal anastomosis (IRA). A segmental resection may result in a better function, but full colonoscopic surveillance is still required due to a risk of further CRC development. Colectomy with IRA reduces this risk. Because the rectum is retained, the morbidity associated with proctectomy is avoided and surveillance made easier. A proctocolectomy, in the form of an end ileostomy or RPC, is recommended for rectal cancers.
10.4 Familial Colorectal Cancer Type X
Lindor et al. introduced this term to describe individuals meeting the Amsterdam criteria with MSI negative colorectal cancers (Lindor et al. 2005). Family members meeting these criteria are at lower risk of developing CRC than in Lynch syndrome but will still require 3–5-yearly colonoscopic surveillance (Dove-Edwin et al. 2006).
10.5 Familial Adenomatous Polyposis (FAP)
FAP is a rare, autosomal, dominantly inherited condition characterised by the development of hundreds to thousands of colorectal adenomas. The population prevalence of FAP is between 1/7,500 and 1/13,000 with almost 100 % disease penetrance by 40 years of age (Bisgaard et al. 1994).
Without prophylactic colectomy, almost all individuals affected will develop CRC by the fourth decade of life (Petersen et al. 1991). Polyps also occur in the stomach and duodenum. Fundic gland polyps in the stomach pose no malignant risk (Wu et al. 1998b). Ninety per cent of those with FAP develop duodenal adenomas, and in 10 % of cases these can become malignant (Wallace and Phillips 1998). They are mostly peri-ampullary and associated with poor prognosis if they progress to cancer (Clark 2009).
Extra-intestinal manifestations of FAP include osteomas, dental abnormalities (e.g. supernumerary teeth), congenital hypertrophy of the retinal pigment epithelium (CHRPE), epidermoid cysts, adrenal adenomas, cancers (including thyroid, central nervous system tumours and hepatoblastoma) and desmoid tumours (Half et al. 2009).
10.5.1 Genetics
FAP is due to germline mutation in one of the copies of the tumour suppressing adenomatous polyposis coli (APC) gene.
10.5.2 Diagnosis
Traditionally, FAP was diagnosed by the presence of more than 100 colorectal adenomas, but this definition fails to incorporate all cases: i.e., in attenuated FAP only 10–100 polyps may occur, and CRC presents at a later age (Hernegger et al. 2002). In up to 80 % of cases, FAP can be confirmed genetically by identifying the APC mutation responsible. Most will originate from previously known FAP families but 20 % arise as a new mutation (Bisgaard et al. 1994).
An expert endoscopist is important in the accurate assessment and diagnosis of FAP. The use of chromoendoscopy (dye spray) can avoid under reporting of polyp burden and misdiagnosis (Wallace et al. 1999). An upper gastrointestinal (GI) endoscopy can be helpful in confirming the diagnosis of FAP as the majority will have duodenal adenomas and fundic gland polyps.
Registries play a fundamental role in ensuring the welfare of people with polyposis syndromes, from identifying at-risk family members through detailed family pedigrees to arranging regular follow-up and offering genetic testing and counselling where appropriate. When genetic testing is not appropriately delivered, there may be adverse consequences to patient care including inaccurate information giving and inadequate counselling (Giardiello et al. 1997). The affected individual should be genetically tested, and where the mutation is identified, other family members can be offered predictive testing; if negative, they can be discharged from follow-up (Berk et al. 1999). Predictive testing is typically offered to children around the age of 12 years as it is rare for advanced colorectal adenomas to develop before this time. If no mutation is identified, then clinical surveillance is required for at-risk individuals.
10.5.3 Surveillance
Annual flexible sigmoidoscopy is recommended from 13 to 15 years but should be undertaken earlier should symptoms develop (e.g. change in bowel function, anaemia or bleeding per rectum). Screening for FAP at a younger age is not recommended due to ethical considerations and because intervention is rarely necessary in the asymptomatic (Tudyka and Clark 2012). Children should be followed up by a specialist paediatric gastroenterologist who will ensure they receive appropriate endoscopic assessment and management. Those developing more or larger polyps than expected for their age may require earlier referral for surgical intervention.
Provided no polyps are seen at flexible sigmoidoscopy, colonoscopy should commence from 20 years of age and continue at 5-yearly intervals with annual flexible sigmoidoscopy in the intervening periods. This algorithm also applies for at-risk individuals where no mutation can be identified.
10.5.4 Surgical Intervention
It is important to appreciate the genotype-phenotype correlation in FAP when planning surgery. The APC gene mutation site can affect phenotypic expression (Wu et al. 1998a). Mutations located between codons 1251 and 1309 predispose to a higher colorectal polyp burden, particularly in the rectum, and cancer often develops earlier. In contrast, mutations at the 3′ and 5′ ends of the gene tend to result in a milder ‘attenuated’ phenotype (Soravia et al. 1998; Nieuwenhuis and Vasen 2007). Some individuals with identical mutations exhibit different phenotypic expression which suggests that environmental factors or other genes are important in disease manifestation. Mutations 3′ of 1399 are associated with a higher risk of desmoid development, which can lead to significant morbidity (Sinha et al. 2010). This knowledge may impact upon surgical decision-making, influencing the timing and type of surgery offered.
Once a diagnosis is confirmed, prophylactic colectomy or proctocolectomy is usually offered. A thorough colonoscopy is required to determine polyp burden prior to surgery. Unless this is high or polyps cause symptoms, surgery can be planned for a convenient time, e.g. during school holidays, to minimise impact on education and other activities. For the majority, surgery will be undertaken around mid to late teens.
Until the late 1970s, the only surgical options were a total proctocolectomy (TPC) with end ileostomy or a colectomy with IRA. 1978 saw the advent of RPC with ileo-anal anastomosis (Parks and Nicholls 1978). More recently, a laparoscopic approach has become a surgical option, with the advantage of improved cosmesis.
10.5.5 Surgical Options
For the majority of people facing surgery, a permanent ileostomy will not be a real consideration, and so the decision regarding the type of surgery will come down to either RPC or colectomy and IRA. The advantages and disadvantages of these procedures are listed in Box 10.3.
10.5.5.1 TPC
The whole of the large bowel is excised and a permanent end ileostomy formed, completely removing the risk of CRC. This will rarely be performed prophylactically but is appropriate for very low rectal cancers.
10.5.5.2 Colectomy and IRA
This can be a laparoscopic or open procedure. Defaecatory frequency and leakage are less when compared to RPC (Aziz et al. 2006). The major disadvantage is the carcinoma risk in the retained rectum, which rises sharply after 50 years of age (Fig. 10.2) (Tudyka and Clark 2012; Nugent and Phillips 1992; Nugent et al. 1993). For those requiring subsequent rectal excision, an ileo-anal pouch may be suitable or otherwise a completion proctectomy with end ileostomy will be necessary.
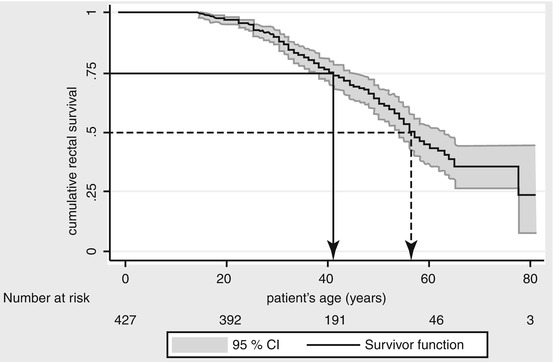
Fig. 10.2
Kaplan-Meier plot showing survival of a healthy rectum (i.e., not requiring removal because of cancer or high adenoma burden) plotted against patient age following IRA. St Mark’s Hospital data 1948–2007. Tudyka and Clark (2012) (Reproduced with permission from Hellenic Society of Gastroenterology)
10.5.5.3 RPC
This operation is an attractive option as nearly the entire large bowel is removed. However, RPC can significantly reduce female fecundity and in 1–2 % of men lead to erectile or ejaculatory dysfunction (Aziz et al. 2006; Cornish et al. 2007). Controversy remains regarding the best technique for anastomosing the pouch and anus. Stapling leaves a small cuff of rectal mucosa where tumours can develop (Van Duijvendijk et al. 1999a). Performing a mucosectomy and a handsewn anastomosis instead is technically challenging, may affect functional outcome and does not abolish cancer risk. Adenomas and carcinomas can also occur in the pouch itself (Church 2005).
10.5.6 Other Considerations and Multidisciplinary Surgical Decision-Making
Historical data found a cumulative rectal cancer risk in the retained rectum after IRA of up to 30 % by 60 years of age. Before RPC, there was no alternative to permanent ileostomy following colectomy other than IRA, so many chose this, regardless of polyp burden. Managing the rectum post-surgery was difficult before flexible endoscopy became available.
It is known that some groups with FAP carry a higher risk of CRC development, e.g. those with codon 1309 mutation. RPC is recommended here rather than IRA, as the risk of developing significant rectal polyposis and requiring proctectomy is high. RPC is also recommended in severe phenotypes, e.g. over 500 colonic adenomas or more than 20 rectal adenomas (Church et al. 2003; Sinha et al. 2010).
It is vital that a multidisciplinary team (MDT) of specialists are involved in the surgical management of patients. In the first instance, it is important that the geneticist can identify the APC mutation where possible. Then, a skilled endoscopist is essential in determining the true extent of polyp burden in the colon and rectum. A pathologist must assess all histology accurately as this may influence the timing of surgery. Specialist nurses and paediatric gastroenterologists often build bonds with patients over years of clinic attendance and may act as patient advocates during MDT discussions. Finally, all available information is presented and discussed within the MDT meeting and surgical recommendations offered to the patient. In cases of attenuated polyposis, surgery may be deferred to a later stage if polyp burden remains small. Some can remain endoscopically manageable with annual surveillance. This may be particularly relevant for those with a desmoid prone mutation in whom trauma from surgery may stimulate desmoid development. Complex cases such as these highlight the importance of MDT discussion and the provision of patient-tailored care.
Patients are counselled about the advantages and disadvantages of each procedure in a specialist clinic setting so they can make an informed decision regarding their surgery. No significant difference has been demonstrated in bowel function and quality of life when comparing RPC and IRA (Aziz et al. 2006; Van Duijvendijk et al. 1999b; Ko et al. 2000; Günther et al. 2003; Hassan et al. 2005). A selective patient-tailored approach results in a better outcome following IRA (Church et al. 2003).
Lifelong follow-up is necessary for individuals with FAP, with endoscopic and clinical examination of the rectum or pouch. After colectomy, the major causes of morbidity and mortality are duodenal cancer and desmoid tumours (Nugent et al. 1993; Bertario et al. 1994), and it is important that these are monitored in a specialist, multidisciplinary setting too.
Box 10.3. Comparing Advantages and Drawbacks of Colectomy and IRA and RPC
Colectomy and IRA | RPC | |
|---|---|---|
Advantages | One-stage procedure, no ileostomy | Less frequent endoscopic surveillance |
Acceptable bowel frequency (×3/day). Incontinence rare | Lowered cancer risk | |
Technically easier to perform laparoscopically/open | ||
Lower perioperative morbidity | ||
No effect on fertility/erectile function | ||
Disadvantages | Intensive endoscopic follow-up | Bowel frequency typically ×5/day. Impaired continence common |
Completion proctectomy sometimes needed | Usually two-stage procedure with temporary ileostomy. Complex surgery | |
Rectal cancer risk (Nugent and Phillips 1992) | Higher perioperative morbidity (Björk et al. 2001)
Related posts: Transdisciplinary Nursing Transdisciplinary Nursing
 Transdisciplinary Management of Perioperative Nutrition Transdisciplinary Management of Perioperative Nutrition
 Team-Based Integrative Care for Recurrent and Locally Advanced Rectal Cancer Surgery Team-Based Integrative Care for Recurrent and Locally Advanced Rectal Cancer Surgery
 Integrating Science and Technology to Proctology Integrating Science and Technology to Proctology
 Metastatic Colon and Rectal Cancer: Role of Multidisciplinary Team-Based Management Metastatic Colon and Rectal Cancer: Role of Multidisciplinary Team-Based Management
 Transdisciplinary Stoma Care Transdisciplinary Stoma Care
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|