DEFINITION OF THE PROBLEM
Local anesthetic systemic toxicity (LAST) may result from unintended intravascular injection of what would have been an appropriate dose of LA (had it been injected in the intended site), or from absorption of LA after peripheral tissue injections (this could arise from unexpectedly rapid absorption of an appropriate dose or absorption at the customary rate of an excessive dose). The end result of all these scenarios is excessive concentrations of LAs at an active site for toxicity. The central nervous and cardiovascular systems (CNS and CVS) are the “toxic” organ targets of increasing LA blood levels. The CNS, being more sensitive to LAST than the CVS, manifests the characteristics of intoxication before the CVS in most circumstances (except in some cases of bupivacaine or etidocaine intoxication).4,5 CNS toxicity is “classically” expressed in two stages. It may begin with an excitation phase (with a progressive sequence of signs and symptoms including shivering, muscle tremors, tonic-clonic seizure activity) due to depression of central inhibitory pathways (so-called disinhibition), which may progress to a depressant phase—for example, hypoventilation and respiratory arrest—due to more global CNS inhibition. The pathophysiology of LA CVS toxicity has both indirect and direct components. With CNS excitation, sympathetic nervous system activation produces tachycardia and hypertension. However, as blood concentrations increase, direct LA-mediated arrhythmogenic and myocardial depressant actions first blunt, then supersede the sympathetic nervous system effects. The resulting arrhythmias, hypotension, and contractile dysfunction contribute to difficulties with resuscitation, altogether defining the LA toxicity phenotype.
 SCOPE OF THE PROBLEM
SCOPE OF THE PROBLEM
Since Albright’s3 report of cardiac arrest after bupivacaine and etidocaine, the incidence of LAST appears to have declined, although the data to document this conclusion are sketchy at best. If true, this improvement could be attributed to better regional anesthesia techniques, greater awareness of the problem, earlier recognition of potential LA toxicity, or reduced use of 0.75% bupivacaine. Findings from preclinical laboratory investigations underlie all of these potential etiologies.6–8 Even so, data from an American Society of Anesthesiology Closed Claims Project show that toxicity from unintentional intravenous (IV) LA injection was the second largest category of regional anesthesia-related claims (behind spinal anesthesia) that resulted in death or brain damage.9 Currently, the incidence of CNS toxicity (seizure) with epidural anesthesia is estimated at 3/10,000 patients and with peripheral nerve blockade the incidence is estimated at 11/10,000 patients.10–12 In Brown’s12 retrospective study of nearly 26,000 patients receiving regional anesthesia, the incidence of seizures from brachial plexus anesthesia was 7.9/1,000 with interscalene or supraclavicular blocks and 1.2/1,000 with axillary blocks (the difference likely arises from the nearly mandatory seizure from injection of any amount of LA into the carotid or vertebral arteries during attempted interscalene block). Taken together with the increasing popularity of nerve block anesthesia among anesthesiologists and surgeons, clinicians are obliged to understand the pathophysiology and management of LAST to ensure favorable clinical outcomes.
 PATHOPHYSIOLOGY
PATHOPHYSIOLOGY
Mechanisms of LA Action
To understand the mechanisms of LAST, it is helpful to review LA mechanisms of action, specifically, interactions between LAs and voltage-gated sodium (Nav) channels. Interactions with Nav channels underlie peripheral nerve block and regional anesthesia. These Nav channels are integral membrane proteins containing one larger α-subunit and one or two smaller β-subunits. The α-subunit, the site of ion conduction and LA binding, consists of roughly 2,000 amino acids arranged in four homologous domains, each with six α-helical membrane-spanning segments.13,14
Nav channels exist in at least three native conformations: resting, open, and inactivated. The neuronal Nav channels “open” briefly during an action potential, which allows extracellular Na ions to flow into the cell and depolarize the plasma membrane. After only a few milliseconds, Nav channels “inactivate” (whereupon the Na current ceases). Local anesthesia and antiarrhythmic actions result when LAs bind Nav channels and inhibit the Na permeability that underlies action potentials in neurons and cardiac myocytes.15,16 The effects of membrane potential (voltage) on this process are complex: membrane potential influences both Nav channel conformations and LA affinity. Membrane depolarization favors first “opening” and then inactivation of the channels. LAs have greater affinity for open or inactivated channels than they do for resting Nav channels. LA inhibition of Na currents increases with repetitive depolarizations, a phenomenon called “use-dependent” or “phasic” block. With successive depolarizations, a declining fraction of Nav channels remain yet “unbound” by an LA, thus the evoked current decline to a nadir.15–17
Aside from Nav channels, LAs will bind to many different sites, including potassium (K) and calcium (Ca) channels, enzymes, N-methyl-D-aspartate receptors, β-adrenergic receptors, and nicotinic acetylcholine receptors. LA binding to these other sites may underlie LA production of toxic side effects,15,16,18 and it is not necessarily true that LAST reactions arise from a mechanism similar to that underlying local anesthesia.
LA Concentrations, Protein Binding, and Metabolism
The likelihood of systemic toxicity increases as the LA concentration in blood increases. Peak LA concentrations vary widely even when the various nerve block techniques are performed properly. Intercostal blocks consistently produce greater peak LA concentrations (after the same LA dose) than plexus or epidural blocks (Fig. 7-1).2,19–21 Addition of epinephrine to LA mixtures will usually reduce peak LA blood concentrations, while the addition of clonidine may increase peak LA blood concentrations. In blood, all LAs are partially protein bound, primarily to α1-acid glycoprotein (AAG) and secondarily to albumin.1,2,19 The more potent, more lipid-soluble LAs have a greater extent of protein binding than the less potent, less lipid-soluble agents. The extent of protein binding of a specific LA is influenced by the plasma concentration of AAG. Both protein binding and protein concentration decline during pregnancy.22 A long-standing assumption in regional anesthesia is that protein binding serves to reduce the potential systemic toxic effects of absorbed or injected LAs. The apparent safety of longer-term epidural infusions of LA and LA-opioid combinations may result in part from progressively increased concentrations of the LA/AAG complexes.23 Other physiochemical properties that influence systemic toxicity are listed in Table 7-1.24
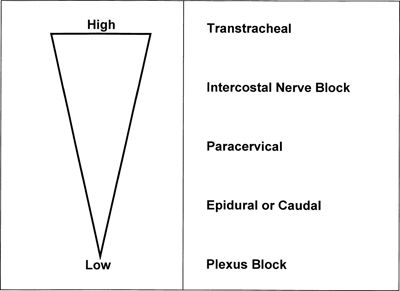
FIGURE 7-1. Route and sites of administration of LAs influence systemic absorption and rate of accumulation. Schematic represents LA plasma concentrations from highest to lowest after the same dose of LA is injected at different sites. (Reprinted from Groban L, Dolinski SY. Differences in cardiac toxicity among ropivacaine, levobupivacaine, bupivacaine, and lidocaine. Tech Reg Anesth Pain Manag 2001;5:48–55, with permission.)
TABLE 7.1 Physiochemical Properties That Influence Systemic Toxicity
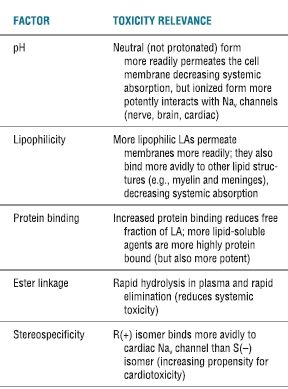
Adapted from Groban L, Dolinski SY. Differences in cardiac toxicity among ropivacaine, levobupivacaine, bupivacaine, and lidocaine. Tech Reg Anesth Pain Manag 2001;5:48–55, with permission.
The latency of LAST onset, particularly related to CNS symptoms, depends on the specific regional technique. As noted previously, seizure activity may occur sooner during placement of an interscalene block than during an epidural, reflecting the inherent differences between an unintentional injection of LA into an artery feeding the brain versus an unintended injection in an epidural vein. Additionally, the time to seizure could be delayed up to 30 minutes after a peripheral nerve block when the seizure is the result of systemic absorption after peripheral injection of a large LA dose.10–12
LA metabolism may also influence the extent and duration of LAST reactions. In general, LAs with greater rates of elimination will have a greater margin of safety. The archetypical example of an agent with a fast rate of elimination is 2-chloroprocaine, and when given to human volunteers, 2-chloroprocaine-induced CNS symptoms abate rapidly.
2-Chloroprocaine and other esters undergo rapid hydrolysis in blood, catalyzed by pseudocholinesterase.1,2,19 Thus procaine and benzocaine are rapidly metabolized to para-aminobenzoic acid (PABA), the compound underlying anaphylaxis to these agents.2 The amides undergo metabolism in the liver.1,2,19 Prilocaine undergoes hydrolysis to o-toluidine, which causes methemoglobinemia in a dose-dependent manner.2,23 For this reason, prilocaine is no longer used in large doses in the United States, and in recent studies benzocaine (used for topical anesthesia) was the most common LA associated with medically important methemoglobinemia in American hospitals.25
CNS Pathophysiology
While CNS manifestations of LAST have historically been described as occurring along a spectrum of clinical findings culminating in seizures (and possibly in CVS collapse if seizures are not aborted), in reality this stereotypical progression of clinical findings occurs only about 60% of the time following increases in LA blood concentrations.26 In a case series of 93 incidents of LAST, the presenting signs were referable to the CNS in 45%, to both the CNS and the CVS in 44%, and to the CVS in the remaining 11%.26
A general relationship exists between the anesthetic potency and CNS LAST. LAs that are more potent at causing conduction blockade produce seizures at lower blood concentrations and lower doses than less potent LAs. In animal studies, there are also age-related considerations, with younger pigs requiring greater doses to produce convulsions than older ones, in animals receiving lidocaine infusions to create EEG-confirmed seizures.27 Among the LAs in the pipecoloxylidide family (including mepivacaine, ropivacaine, and bupivacaine), potency at CNS toxicity is 1.5 to 2.5 times greater for the R(+) isomer as compared to the S(−) isomer. In rabbits, the mean convulsive dose of S(−) bupivacaine was nearly double that of R(+) bupivacaine.6 In sheep, 40 mg R(+) bupivacaine consistently induced convulsions while convulsions never occurred after the same dose of S(−) bupivacaine.28 These stereospecific dose relationships were confirmed in a study by Denson et al.29 in which a more profound depression of the firing rate of cells of the nucleus tractus solitarius occurred with R(+) bupivacaine than with S(−) bupivacaine in anesthetized rats. Racemic bupivacaine and ropivacaine have differing potency at inducing CNS toxicity, although in equipotent concentrations in experimentally induced respiratory acidosis in rats, both agents similarly suppress baroreflex sensitivity, believed to be crucial for maintaining CVS stability.30
The mean convulsant dose of ropivacaine in sheep was 1.33 times that of bupivacaine.31 Likewise, human volunteers tolerated 25% larger IV doses of ropivacaine (124 mg) as compared to bupivacaine (99 mg).32 Knudsen et al.’s33 double-blind crossover study in human volunteers demonstrated that the threshold concentration associated with CNS symptoms was approximately 0.6 mg/L for ropivacaine and 0.3 mg/L for bupivacaine and the time to disappearance of all symptoms was significantly shorter for ropivacaine than bupivacaine. Nevertheless, in rats, the same concentration of bupivacaine and ropivacaine (10 μg/mL) inhibited hippocampal field potentials in the CA1 region. These inhibitory effects on hippocampal field potential amplitude and recovery rate following washout may represent the underlying mechanism for CNS-related LAST.34
There have also been comparisons between S(−) isomers. The relative potency of levobupivacaine and ropivacaine at CNS toxicity varies among species.35–37 In the anesthetized, ventilated rat, Ohmura et al.35 showed that cumulative convulsive doses were similar for levobupivacaine and ropivacaine. In conscious sheep, the convulsant dose was slightly greater for ropivacaine than for levobupivacaine.36,37 When levobupivacaine and ropivacaine were administered to human volunteers in a double-blind randomized crossover trial, early CNS toxic symptoms occurred after equal concentrations, equal milligram doses, and equal infusion rates38 (Box 7-1).
BOX 7-1 Factors Influencing the Threshold LA Dose to Induce Convulsions
 Site of injection
Site of injection
 Rate of injection
Rate of injection
 Rate at which blood concentrations increase
Rate at which blood concentrations increase
 Acidosis
Acidosis
 Level of sedation
Level of sedation
 Stereospecificity of LA
Stereospecificity of LA
Cardiovascular Pathophysiology
The CVS is affected at all stages of LA systemic toxicity. In early stages when CNS symptoms dominate the clinical picture, CV toxicity occurs indirectly (due to CNS excitation) rather than a direct effect on myocardium. This is typically observed in situations in which the toxicity is due to an LA with a greater CV/CNS toxicity ratio, for example, lidocaine, or the route of LA administration results in a gradual systemic absorption. Ladd et al.39 have elegantly demonstrated the actual CV pathophysiology during the CNS excitation phase. In their study, carotid injection of LAs in awake ewes at a dose range that resulted in CNS symptoms (hypertonia, seizures) without arrhythmias or cardiac arrest resulted in hypertension, tachycardia, and a decrease in stroke volume. On the other hand, if the LA being used has a low CV/CNS ratio, signs related to direct cardiac toxicity, for example, arrhythmias and hypotension, might occur first when there is a rapid increase in plasma concentration. This might be a common scenario as a review showed that 55% of cases with LAST presented with CV signs and symptoms (alone or CV and CNS signs together at presentation).26
Direct cardiac toxicity is a complex phenomenon at the molecular level.40 LAs bind and inhibit cardiac Na channels much as they do in neural tissue.15,41–46 In addition, LAs block potassium channels on the cell membrane, prolonging action potential and contributing the cardiotoxicty.43 Calcium channels at the plasma membrane as well as the sarcoplasmic reticulum are also targeted, decreasing the intracellular Ca2+ release and thereby depressing contractility.44–46 Binding and inhibition of the cardiac Ca and K channels by LAs generally occur at concentrations greater than those at which binding to Na channels is maximal.5,15,47 Prolongation of the action potential via the inhibition of Na and K channels and blockage of Ca2+ channels at multiple locations are classically thought to be the major molecular mechanisms behind the major clinical findings of LA-related cardiac toxicity.48 However, metabotropic cellular signaling systems, for example, beta adrenergic and lysophosphatidyl, also appear to contribute to the phenotype of LA-induced cardiac toxicity. Recent observations suggest that inhibition of mitochondrial metabolism and oxidative phosphorylation might also play a role in severe cardiac toxicity.49
Physicochemical properties of the LAs can explain at the molecular level the variation in their potential for causing clinical toxicity. Lipophilicity, anesthetic potency, and the proclivity for protein binding covary with toxicity. For instance, bupivacaine, the canonical cardiotoxic LA, is more lipophilic and more highly protein bound than lidocaine. It also binds more avidly and longer than lidocaine to cardiac Na channels.50 Stereoselectivity is another important factor in determining toxicity.51 The R(+) enantiomer of bupivacaine binds cardiac Na channels more avidly than the S(−) enantiomer, and this finding led to the clinical development of the enantiomeric formulations, levobupivacaine, and ropivacaine.52 As a general rule, LAs inhibit conduction in the heart, with the same rank order of potency as for nerve block.53–55
Similarly, LAs bind β-adrenergic receptors and inhibit epinephrine-stimulated cyclic adenosine monophosphate (AMP) formation, with the same rank order as for LA potency at peripheral nerve block sites. Inadequate signaling by cAMP (by either mechanism) could underlie the refractoriness of bupivacaine CV toxicity to standard resuscitation measures.20,21,56,57 Bupivacaine is also more effective than other less toxic LAs at collapsing the mitochondrial chemiosmotic gradient needed for oxidative phosphorylation. It is also a very potent inhibitor of other enzymes at the inner mitochondrial membrane, such as those necessary for uptake of fatty acids, the heart’s preferred fuel under normal aerobic metabolism. A description of the potential sites at which LAs interfere with cardiac function is displayed in Figure 7-2.
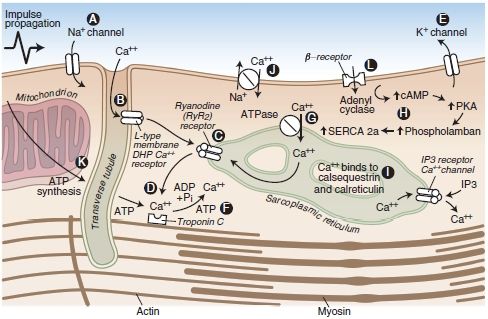
FIGURE 7-2. Cardiac myocyte transport processes and potential sites of LA-induced cardiac toxicity (A-L). LAs block cardiac Na+ channels (A). Bupivacaine displays fast-in, slow-out kinetics, whereas lidocaine displays fast-in, fast-out kinetics within the Na+ channels of the myocardium. The net effect is inhibition of Na+ conductance, leading to a slowed conduction block that may be associated with reduced myocardial contractility. At high concentrations, LAs bind and inhibit cardiac K+ (E) and Ca2+ (B) channels as well as beta-adrenoreceptors. Blockade of cardiac K+ channels (E) may also contribute to the cardiotoxic effects of LAs by lengthening the cardiac action potential, predisposing the heart to ventricular arrhythmias. LAs have a depressant effect on the Ca2+ slow channels (B), attenuating the influx of calcium, which is a vital link to the cardiac excitation-contraction coupling process. LAs may also depress myocardial contraction by interfering with Ca2+ release from the sarcoplasmic reticulum (C). Furthermore, LAs bind and inhibit beta-adrenoreceptors (L) and adenyl cyclase (H), inhibiting cyclic AMP formation, which may impair resuscitative efforts after cardiotoxicity. Finally, an LA-induced interference with mitochondrial energy metabolism can contribute to the electrical and mechanical depression of the myocardium (D, F, G, K). However, these additional mechanisms of cardiotoxicity may require LA concentrations that are greater than the concentrations that would have produced cardiac standstill from block of cardiac Na+ and Ca2+ channels. Each of the essential components of mitochondrial metabolism required for oxidative phosphorylation and synthesis of adenosine triphosphate is impaired by lipophilic LAs. These include transport of fatty acids into the mitochondrial matrix, generation of the chemiosmotic gradient, electron transport, and ATP synthesis.
(Adapted from Groban L, Dolinski SY. Differences in cardiac toxicity among ropivacaine, levobupivacaine, bupivacaine, and lidocaine. Tech Reg Anesth Pain Manag 2001;5:48–55, with permission.)
Arrhythmogenicity, myocardial contractility, and survivability (ease of resuscitation) are the most commonly used endpoints to investigate LA cardiotoxicity in experimental models using intact animals. Nevertheless, there is a great deal of variability in such laboratory studies with respect to the species used as well as experimental design, metrics, and endpoints. There is no consensus as to which model and design most faithfully represent the cardiovascular toxicity from LAs in humans.40
Arrhythmogenicity
The overall direct effects of LAs on cardiac rhythm are depression of excitability and delay in conduction.58 As stated above, when the plasma levels of the LA in question are not sufficiently high, tachyarrhythmias could indirectly result from CNS excitation. At higher plasma concentrations of LA, a dose-dependent prolongation of cardiac conduction is reflected by increases in the PR interval and QRS duration of the electrocardiogram.59–61 At higher doses still, depression of sinoatrial and atrioventricular nodal activity is manifested by bradycardia and atrioventricular block, potentially leading to asystole.60 With increased blood concentrations of bupivacaine, the heart becomes increasingly susceptible to prolongation of the QT interval, ventricular tachycardia, torsades, and ventricular fibrillation.62,63
Bupivacaine is consistently the most arrhythmogenic LA, regardless of the animal species or experimental protocol. At subconvulsant doses, bupivacaine induced nodal and ventricular arrhythmias, whereas even convulsant lidocaine doses do not induce arrhythmias.64 Feldman et al. found that at twice the convulsive dose (with no resuscitative attempts), 83% of bupivacaine-treated dogs died as compared to 17% of ropivacaine-treated dogs.65,66 However, with early resuscitation, mortality decreased from 83% to 33% in the bupivacaine-treated group and from 17% to 0% in the ropivacaine-treated group. Kotelko et al.67 reported that in conscious sheep, convulsant bupivacaine doses produced severe arrhythmias whereas convulsant doses of lidocaine produced only transient ST-segment depression or sinus tachycardia.
Ropivacaine is apparently less arrhythmogenic than bupivacaine. In a crossover experimental design, Knudsen et al.68 found that ropivacaine caused a smaller increase in QRS duration compared to bupivacaine in human volunteers. Since QRS duration have been positively correlated with cardiotoxicity of LAs,53 these findings provide valuable comparison of the cardiotoxicity of ropivacaine and bupivacaine in humans. A study by Reiz et al.54 in pigs comparing bupivacaine, ropivacaine, and lidocaine supports this finding. The endpoint being QRS prolongation, the electrophysiologic toxicity ratio was 15:6.7:1 for bupivacaine:ropivacaine:lidocaine.54
Levobupivacaine has an intermediate arrhythmogenic risk between ropivacaine and bupivacaine.69 In nonanesthetized rats, levobupivacaine prolonged the QRS more than ropivacaine. Ventricular tachycardia occurred in seven of eight rats treated with levobupivacaine compared with only one of eight rats treated with ropivacaine.69 Similarly, in anesthetized rats, the cumulative dose and plasma concentrations of LA at the onset of the first arrhythmia were greater for ropivacaine as compared to levobupivacaine, and both were significantly greater than racemic bupivacaine.69
Arrhythmogenicity of a given LA may be different from its LA potency. For peripheral nerve blocks and epidural anesthesia, bupivacaine is about four times more potent than lidocaine (potency ratio of 1:4).70,71 However, in anesthetized pigs, intracoronary injection of LA produced comparable prolongation of the QRS interval with bupivacaine and lidocaine at a dose ratio of 1:16.72 Moreover, seven of 15 animals given 4 mg of intracoronary bupivacaine died of ventricular fibrillation, whereas lidocaine-induced ventricular fibrillation occurred at 64 mg. Therefore, it can be concluded that while bupivacaine is four times more potent than lidocaine as an LA, it is 16 times more arrhythmogenic.
Administering LA directly in the coronary artery controls for indirect CNS effects, and such studies indicate that severe bupivacaine-induced ventricular arrhythmias result largely from direct cardiac action, and not CNS activation.73 Chang et al.73 studied nonanesthetized sheep receiving intracoronary bupivacaine, levobupivacaine, or ropivacaine. All the three drugs prolonged the QRS duration, but bupivacaine was more potent and produced greater prolongation than ropivacaine. In conscious sheep, IV levobupivacaine produced fewer arrhythmias than bupivacaine.74,75 Bupivacaine doses of 125 to 200 mg produced fatal ventricular fibrillation in some sheep, whereas, similar life-threatening arrhythmias were not found with levobupivacaine doses <225 mg.
Decreased Myocardial Contractility
Dose-dependent reduction in contractility occurs with systemic LA intoxication and unquestionably contributes to LA-induced cardiovascular instability. The rank order for LA potency as negative inotropes is the same as that for their potency at nerve conduction blockade. More potent LAs (e.g., bupivacaine) tend to reduce cardiac contractility at lower doses and concentrations than the less potent LA agents (e.g., lidocaine).67,72 However, comparisons of contractile depression among ropivacaine, levobupivacaine, bupivacaine, and lidocaine are not entirely straightforward. Subconvulsant doses of levobupivacaine and bupivacaine given to nonanesthetized sheep produced comparable contractile depression.75 In sheep, intracoronary ropivacaine produced smaller reductions in myocardial contractility and stroke volume than equivalent doses of bupivacaine or levobupivacaine.73 Likewise, there was no difference between levobupivacaine and bupivacaine. During incremental LA intoxication in anesthetized dogs, ropivacaine produced less left-ventricular depression than levobupivacaine or racemic bupivacaine (Table 7-2). Similarly, myocardial contractility in nonanesthetized sheep was most severely depressed by bupivacaine, followed by levobupivacaine, and ropivacaine (in decreasing order).73
TABLE 7.2 Concentrations of LAs Inhibiting Myocardial Function
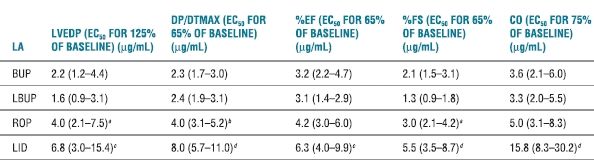
Note: Data represented are EC estimates and 95% confidence intervals.
aROP > LBUP; p < .05.
bROP > BUP, LBUP; p < .05.
cLID > BUP, LBUP; p < .01.
dLID > BUP, LBUP, ROP; p < .01.
EC50, effective concentration for 50% of population; BUP, bupivacaine; LBUP, levobupivacaine; ROP, ropivacaine; LID, lidocaine; base, baseline.
Reprinted from Groban L, Dolinski SY. Differences in cardiac toxicity among ropivacaine, levobupivacaine, bupivacaine, and lidocaine. Tech Reg Anesth Pain Manag 2001;5:48–55, with permission.



