I. GLUCOSE HOMEOSTASIS, INSULIN RESISTANCE, AND INSULIN DEFICIENCY
A. Normal Blood Glucose Dynamics. In the normal fasting state, blood glucose (BG) is regulated between 70 and 110 mg/dL and does not increase to more than 200 mg/dL despite significant fluxes of glucose into the bloodstream after meals. Absorption of a typical meal may require 150 g of glucose (a 30-fold excess of the steady-state amount in the blood) to move through the circulation and into storage within a few hours. A rise in BG of twofold (more than 200 mg/dL) during this movement is abnormal and is sufficient to diagnose diabetes mellitus (DM) in the outpatient setting. Glycated hemoglobin (HbA1c) can be used to evaluate average plasma glucose concentration over an interval of several months.
B. Endocrine Control of Blood Glucose. Pancreatic β-cells secrete insulin directly into the portal circulation in response to the BG level. Glucose is converted to glycogen for storage in the liver and muscle and to triglycerides for storage in the adipose tissue. During fasting, pancreatic β-cells secrete glucagon to promote breakdown of glycogen stores and release of glucose into the blood. Glucagon is also the first line of defense against hypoglycemia.
C. Insulin Resistance and Deficiency. Insulin stimulates glucose uptake and promotes cell growth and survival. In states of insulin resistance, such as type 2 DM and critical illness, much higher levels of insulin can be required for the same degree of glucose uptake. Postreceptor signaling events can be inhibited by the counterregulatory hormones glucagon, epinephrine, norepinephrine, cortisol, and growth hormone, as well as inflammatory cytokines and intracellular free fatty acids. These same counterregulatory hormones can stimulate glycogen breakdown, glucose production from amino acids, and release of fatty acids from lipids. If pancreatic β-cells cannot sufficiently increase insulin production in response to insulin resistance, relative insulin deficiency leads to hyperglycemia. Toxicity of cytokines and hyperglycemia itself can lead to β-cell failure and absolute insulin deficiency superimposed on insulin resistance.
II. HYPERGLYCEMIA OF CRITICAL ILLNESS
A. Pathophysiology. Critically ill patients without preexisting DM frequently become insulin resistant and hyperglycemic due to elevated levels of cytokines such as IL-1, IL-6, and TNF and stress hormones such as cortisol, glucagon, and adrenergic hormones. Treatment with glucocorticoids and sympathomimetic drugs, increased nutrition to compensate for a catabolic state, and administration of intravenous (IV) dextrose all contribute to hyperglycemia. Patients with preexisting DM have insulin resistance and almost always need higher levels of insulin to maintain normal glucose levels when they are critically ill.
B. Hyperglycemia and Outcomes. Hyperglycemia appears to be a marker for severity of illness and is associated with poor outcomes including increased infarct volume after stroke, decreased cardiac function after myocardial infarction, increased wound complications after heart surgery, and increased mortality.
1. While hyperglycemia attributed to critical illness is associated with worse outcomes, hyperglycemia attributed to preexisting diabetes is also associated with increased mortality. In one study, preoperative hyperglycemia (>200 g/dL) was associated with a twofold increase in 30-day mortality and a fourfold increase in 30-day cardiovascular mortality. Studies also suggest that diabetic patients in particular have an increased risk of wound infections, renal failure, and rehospitalization following heart surgery.
C. Intensive Insulin Therapy (IIT). A series of clinical trials that started in 2001 have tested the efficacy of using IV insulin to tightly regulate BG to normal or near normal, a much narrower range than had been customary in the ICU. The trial led by Van den Berghe targeted a blood-glucose level of 80 to 110 g/dL and reduced in-hospital mortality of surgical ICU patients by as much as 34%, as well as reducing the incidence of acute renal failure, bloodstream infections, critical illness polyneuropathy, and the duration of mechanical ventilation and of ICU stay. However, follow-up studies noted the increased risk of severe hypoglycemia and some studies even required early termination due to increased risks in the IIT group. The GluControl and VISEP trials both noted no difference in mortality between tight versus conventional glucose control. In 2009, the NICE-SUGAR trial randomized medical and surgical ICU patients to glucose ranges of 80 to 110 versus 140 to 180 and found a small increase in mortality with intensive glucose control with a number needed to harm of 38. Furthermore, there was no difference in median number of ICU days, duration of mechanical ventilation, or need for renal replacement therapy. Retrospective analysis seems to suggest that the mortality difference could be attributed to whether a patient has preexisting diabetes. Nondiabetics who develop hyperglycemia in the acute setting seem to be at greater mortality risk, although it is unclear whether intensive glucose control seems to favor one population over another.
1. The reason for such wide discrepancy among large, controlled clinical trials is not entirely clear. Confounding factors may include methodology, the different ranges of BG goals tested, preexisting diabetes, the proportions of medical versus surgical patients, and the different provision of total calories as well as their source (enteral vs. parenteral). Although not proven, it may also be that the benefits of IIT derive not only from glucose control, but also from the anabolic effects of insulin. Insulin has been shown to improve protein synthesis, stimulate anti-inflammatory effects, and modulate energy usage.
D. Risks of IIT. The primary risk of IIT is hypoglycemia. Throughout the various IIT trials, severe hypoglycemia (BG <40 mg/dL) occurred in 5% to 19% of study patients and was in some cases identified as an independent risk factor for death in patients treated with IIT. Severe hypoglycemia may cause neurologic damage because the brain depends on glucose. The risk of hypoglycemia to other organs systems is not as well defined, but hypoglycemia due to excess insulin is also associated with low blood levels of free fatty acids, the preferred fuel for the heart. This may be particularly problematic because of increased cardiac demand during critical illness. Intensive glucose control in poorly controlled diabetics may stimulate the body to cause a hypoglycemic stress response.
1. The consequences of hypoglycemia may counteract some or all of the benefits of IIT. In the ICU, the most common cause of hypoglycemia is interruption of nutrition without stopping the insulin infusion. This can occur due to occlusion of feeding tubes, depletion of TPN or IV glucose bags, or accidental removal of tubes or lines. The BG level can fall very rapidly when feeding is interrupted, requiring a high degree of vigilance from staff. If enteral feeding or TPN is interrupted, a D10 infusion should be started immediately and the rate of insulin infusion decreased to prevent a precipitous fall in blood glucose (see Chapter 11).
E. Implementation of Insulin Therapy. To reduce the risk of hypoglycemia and achieve adequate BG control, most ICU protocols specify point-of-care testing of BG every 1 to 2 hours, although this approach requires up to 2 hours of nursing time each day. Given the lack of uniformity of the results of the IIT trials, a wider target range for blood glucose control may confer most of the potential benefit with less risk of hypoglycemia. The 2012 international guidelines from the Surviving Sepsis Campaign sponsored by a number of critical care societies suggest a few main points in implementing IIT:
1. Following initial stabilization, hyperglycemia (BG >180 mg/dL) should be treated with insulin.
2. All patients receiving IV insulin should have BG monitored as frequently as every 1 to 2 hours until BG values are stable, then every 4 hours.
3. Low BG levels obtained from capillary blood by point-of-care testing should be confirmed with a full blood or plasma sample, as the former may not be accurate.
a. As a result, while glucose control in the ICU remains a priority, intensive glucose control to normoglycemic levels is no longer advised. Based on current evidence, targeting a glucose level of <180 mg/dL seems advisable.
F. Dosing Algorithms for Insulin. No consensus has emerged on the best algorithms to use for control of BG with IV insulin, and many forms of paper-based algorithms have been used. There has been interest in developing automated “closed-loop” systems for glucose regulation in the ICU to improve quality of BG control and safety. When such a system becomes available, it will be much easier to conduct the studies required to resolve the many remaining uncertainties about IIT. Very high doses of insulin may be required to control BG in critically ill patients. Insulin infusion rates of greater than 10 U/h are not uncommon and some patients require rates in excess of 50 U/h.
G. Appropriate Use of Insulin Dosing Algorithms. Protocols are not appropriate for all hyperglycemic patients. They are not appropriate for treatment of patients with type 1 DM who need a basal rate of insulin administration to prevent lipolysis and ketone body formation. Protocols for critical illness are also not appropriate for treatment of diabetic ketoacidosis (DKA) and hyperosmolar hyperglycemic states (HHSs) where the primarily goals are closure of the anion gap and normalization of the serum osmolarity, respectively (see below).
H. Transition from IV to Subcutaneous (SC) Insulin. The transition from IV to SC insulin injections requires careful attention. The first dose of a long-acting analog of insulin (usually NPH insulin, twice daily) should be given at least 2 hours prior to discontinuation of the infusion. The total daily dose of long-acting insulin (divided into two daily doses if using NPH insulin) should be at least half of the total insulin dose administered IV over the last 24 hours. The remainder of the insulin requirement may be provided with a sliding scale of regular or rapid-acting formulation (insulin aspart, lispro, or glulisine). A sliding scale order alone, without any standing insulin, is not sufficient and will result in recurrent hyperglycemia. The dose of long-acting insulin should be reevaluated every day. A useful rule of thumb is to add at least half of the sliding scale given on the previous day to the basal insulin dosing for the upcoming day and repeat the process until all or most of the blood glucose values are in the desired range. Conversely, hypoglycemia should usually prompt reduction in the basal insulin dose unless it occurred after a large sliding scale bolus to treat hyperglycemia. All type 1 diabetic patients require basal insulin, whether or not they are eating, to prevent development of DKA. A modest reduction from the home basal insulin dose may be appropriate upon hospital admission depending on the form of basal insulin and tightness of control.
III. DIABETIC KETOACIDOSIS
A. Pathophysiology. DKA occurs due to a lack of insulin or severe insulin deficiency. Clinical states often involve extreme stress, such as infection, myocardial infarction, or postoperative inflammation. In addition to insulin deficiency, DKA is also characterized by an excess of counterregulatory hormones or inflammatory cytokines. The downstream effects include hyperglycemia, lipolysis, ketogenesis, and hyperosmolarity. Hyperkalemia is common despite whole-body potassium depletion because insulin in an important mediator of potassium uptake into cells. Glucosuria results in an osmotic diuresis with prominent potassium and phosphate wasting. There is loss of water in excess of sodium resulting in dehydration and volume depletion. Decreased peripheral vascular resistance, nausea, vomiting, and abdominal pain are likely due to elevated levels of prostaglandins.
B. Symptoms of DKA. Symptoms may include polyuria, polydipsia, polyphagia, weight loss, vomiting, abdominal pain, dehydration, weakness, confusion, or coma. The examination may find poor skin turgor, ileus, Kussmaul respirations (very deep breaths without tachypnea), tachycardia, hypotension, the fruity aroma of ketones on the breath, coffee-ground emesis (hemorrhagic gastritis), altered mental status, shock, and coma. The patient may have a warm and well-perfused periphery despite severe volume depletion. Full-blown DKA can evolve in less than 24 hours.
C. Causes of DKA. DKA occurs when insulin is mistakenly withheld or reduced, when insulin is inactive, or when an acute illness increases insulin requirements. Patients with type 1 DM are most at risk but patients with type 2 DM can also develop DKA in the setting of a catastrophic illness or as the initial presentation of the disease. Newly diagnosed type 1 diabetics account for 15% of DKA. The possibility of DKA should be considered in any patient with diabetes and critical illness. Drugs can uncommonly precipitate DKA. A partial list of causes for DKA is shown in Table 27.1.
D. Diagnosis of DKA. Diagnosis requires the presence of serum ketones and is supported by anion gap >12 mEq/L (AG = Na − [Cl + HCO3]), plasma glucose >250 mg/dL, pH <7.3, serum bicarbonate <18 mEq/L, and moderate to large ketones in the urine. Patients who have been able to keep up with volume and water losses by drinking may have been able to excrete enough ketones as sodium salts with retention of chloride so that they present with a hyperchloremic nonanion gap metabolic acidosis. Treatment of DKA with extra insulin by the patient or another provider can decrease BG to <200 mg/dL without clearing ketones to produce “normoglycemic DKA.” See Table 27.2 for the differential diagnosis of anion gap acidosis. See Figure 27.1 for summary of diagnosis and management of hyperglycemic emergencies.
| Causes of Diabetic Ketoacidosis | |
• Omission of insulin, inappropriate reduction in dose, inadvertent use of denatured insulin (insulin exposed to heat)
• Infection/sepsis
• Infarction
• MI, bowel ischemia, stroke
• Endocrine abnormalities:
• Pheochromocytoma
• Acromegaly
• Thyrotoxicosis
• Glucagonoma
• Pancreatectomy
• Medications/drugs:
• Ethanol abuse (DKA can be confused with alcoholic ketosis)
• Atypical antipsychotics—olanzapine, clozapine, risperidone
• Anticalcineurin drugs—FK506
• HIV protease inhibitors
• α-Interferon/ribavirin therapy
• Corticosteroids
• Sympathomimetics (cocaine, terbutaline, dobutamine)
• Pentamidine
• Thiazides
• Other conditions that may predispose to DKA:
• Pancreatitis (reduced insulin secretion and insulin resistance)
• Surgery
• Trauma
• Pregnancy
• Eating disorder
| Differential Diagnosis of Anion Gap Acidosis | |
• Starvation ketosis—bicarbonate rarely <18, no hyperglycemia
• Alcoholic ketoacidosis—glucose usually <250, may be hypoglycemic
• Lactic acidosis—serum lactate
• Renal failure—BUN, Cr (note that the measured Cr can be artifactually elevated by acetoacetate depending on the assay used)
• Salicylate intoxication—salicylate level
• Methanol—methanol level
• Ethylene glycol—calcium oxylate and hippurate crystals in urine
• Paraldehyde ingestion—usually hyperchloremic, strong odor on breath
E. Measurement of Serum Ketones. Measurement of serum ketones with traditional methods such as the nitroprusside test may initially underestimate or overestimate ketone levels. Treatment may increase the apparent levels of ketones in the blood by converting a poorly detected form (β-hydroxybutyrate) to a more easily detected form (acetoacetate). Therefore, ketone levels should be used to make the diagnosis, but not to follow the progress of therapy.
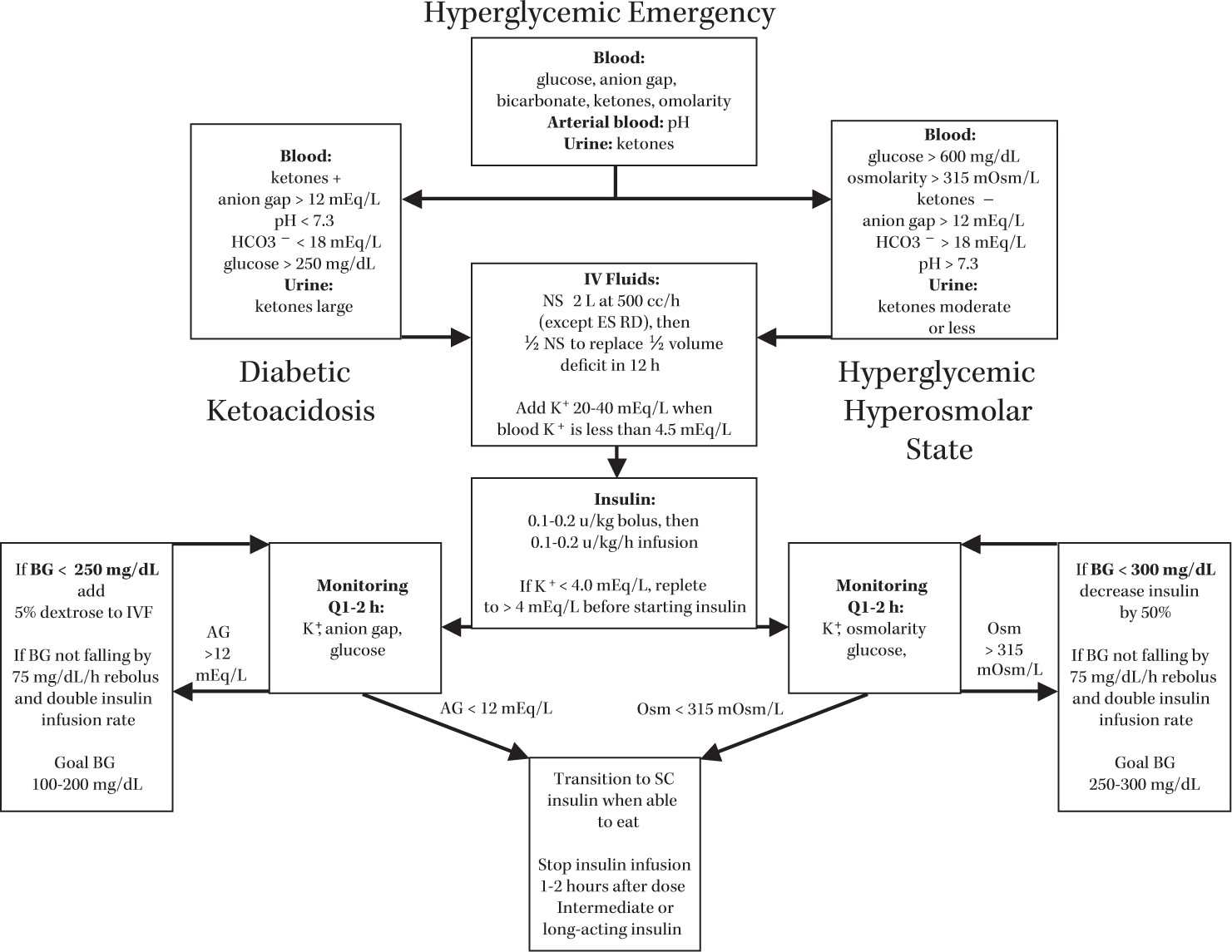
FIGURE 27.1 Diagnosis and management of hyperglycemic emergencies. Not all elements are required for diagnosis—key elements for DKA are blood ketones and anion gap >12, for HHS BG >600 and Osm >315 mOsm/L.
F. Goals of Therapy and Search for the Cause of DKA. Normalization of glucose is not sufficient treatment of DKA. The primary goals of therapy are to treat hypovolemia, to normalize blood potassium concentration and replete potassium stores, to close the anion gap, and to identify and treat the underlying cause of DKA. Even if insulin omission is suspected, a full workup for other causes is mandatory. Initial evaluation should include a comprehensive set of serum electrolytes, but strong consideration should also be given to cultures to rule out infection. CXR, ECG, and CT should be considered to find a precipitating cause. Certain laboratory anomalies are common in DKA, which may obscure the underlying cause. Amylase and lipase elevations of less than threefold are not sufficient to diagnose pancreatitis in DKA. Leukocytosis with elevated PMNs may be due to stress and proportional to ketonemia, or secondary to underlying infection. Creatinine is often elevated due to hypovolemia (with elevated BUN/Cr ratio), but ketone bodies may also interfere with the creatinine assay. Liver enzymes may be elevated. All of these abnormalities should resolve with treatment of DKA if there is not another underlying cause.
G. Volume Repletion. Typical volume deficits in DKA are 10% of body mass. Fluid repletion should begin with approximately 2 L of NS administered at a rate of 1,000 mL/h. After the initial hydration, fluids should change to ½ NS, assuming the corrected serum sodium (taking serum glucose into account) is normal or high. The rate should be reduced with a goal of correcting half of the volume deficit in the first 12 hours and the remainder in the next 12 hours. More rapid administration of fluid may delay resolution of acidemia by diluting bicarbonate. In children, too rapid repletion of volume has been associated with cerebral edema, which can be fatal. In adults, excess administration of NS can delay resolution of the hyperchloremic metabolic acidosis, which may underlie or follow treatment of the anion gap acidosis. End-stage renal failure patients who are anuric are a special case and are likely to need little or no IV fluid, as they are not capable of osmotic diuresis in response to hyperglycemia.
H. Potassium Repletion. Potassium (K+) depletion is almost universal in DKA (with the exception of oliguric or anuric patients) with a typical deficit of 3 to 5 mEq K+ per kilogram body weight that requires aggressive repletion. Glucosuria can result in the loss of 70 mEq of potassium for each liter of volume lost. The initial serum K is often elevated despite severe total body depletion due to shifts from within cells to the extracellular fluid. Insulin administration will lower the potassium concentration in the blood. Potassium should be added at 20 to 40 mEq/L to the IVF when the serum K+ is 4.5 or less. The serum K+ should be followed closely and repleted aggressively with cardiac monitoring to a level of 4 to 5 mEq/L. If the patient is hypokalemic on presentation, potassium should be repleted to the lower limit of normal before insulin is given to avoid severe hypokalemia and life-threatening arrhythmias.
I. Insulin Therapy. Insulin is given IV with an initial bolus of 0.1 to 0.2 U/kg (or 10 U) followed by a continuous IV infusion of approximately 0.1 U/kg/h (or 10 U/h). Insulin is typically mixed at 1 U/mL. Lower concentrations may lead to proportionally large losses by adherence to the infusion bag or IV tubing. In-line filters may bind and deplete insulin. The combined effect of insulin effect and fluid repletion will usually lead to a decrease in the blood glucose of 75 to 100 mg/dL/h. If the decrease in BG is less than 75 mg/dL/h, the insulin dose may be doubled. If the decline is still too slow, a new bottle of insulin should be used.
J. Closure of the Anion Gap. The endpoint of therapy should be closure of the anion gap (<12 mEq/L) and resolution of the acidosis (pH >7.3, bicarbonate >18 mEq/L). If glucose falls to less than 250 mg/dL but the anion gap is not closed, 5% dextrose should be added to the IV fluids (e.g., D5 ½ NS at 100 cm3/h) and the insulin dose should be decreased to 0.05 to 0.1 U/kg/h (or 5 U/h). The insulin dose should then be adjusted as needed to hold the glucose at 100 to 200 mg/dL until the anion gap is closed. Measurement of ketones should not be used to determine the endpoint of therapy. It is not necessary to eliminate ketonemia and ketonuria as long as the anion gap is closed. A hyperchloremic, nonanion gap acidosis is a very common consequence of aggressive hydration with saline. It does not need to be treated and will correct itself over several days by renal excretion of ammonium chloride if renal function is adequate.
K. Phosphorus and Bicarbonate. Phosphate is often normal to high in DKA, but falls with treatment. Phosphorus repletion is only indicated in patients with phosphorous less than 1.0 mEq/L and with a clinical syndrome consistent with hypophosphatemia, which may include hemolytic anemia, platelet dysfunction with petechial hemorrhage, rhabdomyolysis, encephalopathy, seizure, heart failure, and weakness of respiratory or skeletal muscles. There is no benefit of bicarbonate treatment in patients with pH greater than 6.9. Administration of insulin will result in utilization of ketones in the TCA cycle and regeneration of bicarbonate. Extra bicarbonate can increase potassium requirements, may increase hepatic production of ketones, and may delay resolution of cerebral acidosis.
L. Complications of DKA. The most feared complication of DKA is cerebral edema, which occurs in up to 1% of children with DKA, but rarely in adults. However, the mortality in cerebral edema can be as high as 24%. Cerebral edema as a complication of DKA is a diagnosis of exclusion in adults, and other causes of depressed mental status should be carefully investigated.
M. Transition from IV to SC insulin (see Section II.H) should begin when the patient is able to eat. A dose of the long-acting insulin (NPH, glargine, or detemir) should be given or continuous SC infusion should be started 1 to 2 hours before the insulin infusion is stopped. In cases of uncomplicated DKA (i.e., due to insulin omission), the patient’s home regimen can be restarted if home control (as judged by the HgbA1c) was adequate. Making the transition to SC insulin in the morning or evening is strongly preferred so that there is a smooth transition to the patient’s usual insulin schedule.
IV. HYPEROSMOLAR HYPERGLYCEMIC STATE
A. Pathophysiology. Insulin is able to suppress lipolysis and ketogenesis at much lower concentrations than is required to stimulate glucose uptake. In contrast to patients with type 1 DM, patients with type 2 DM usually have sufficient insulin production to prevent the excessive production of ketones. When severe hyperglycemia occurs in the absence of ketosis, the syndrome is called HHS or hyperosmolar nonketotic coma (HONKC). BG may rise to levels that are unusual in DKA, often greater than 1,000 mg/dL. The blood pH and bicarbonate levels are typically normal and tests for ketones are negative. Both HHS and DKA are associated with hyperosmolarity, polyuria, polydipsia, volume depletion, and whole-body potassium depletion, although the serum potassium is usually normal or elevated before treatment. The two syndromes are part of a spectrum and approximately one-third of patients have some aspects of both syndromes. Altered mental status, including obtundation and coma, are more common in HHS because the degree of hyperglycemia and hyperosmolarity are higher. Patients with HHS may also develop seizures or focal neurologic signs, and up to 50% of patients present with coma. As in DKA, patients with oliguric renal failure have a different presentation. Although glucose levels are high, the serum sodium is reduced to compensate so that there is minimal hyperosmolarity and few neurologic symptoms.
B. Diagnosis. The diagnosis of HHS requires severe hyperglycemia (>600 mg/dL, often >1,000 mg/dL) and hyperosmolarity without an anion gap acidosis. The American Diabetes Association categorizes HHS with a serum osm of >320 mOsm/kg and a pH >7.3. HHS exists on a spectrum with DKA and some patients with HHS may have modest ketonemia, while some patients with DKA may have more severe hyperosmolarity than is typical of DKA. See Figure 27.1 for summary of diagnosis and management of hyperglycemic emergencies.
C. Treatment. IV fluid replacement and insulin therapy are the mainstays of treatment for HHS, just as they are for DKA, but the goals of insulin therapy differ. Fluid replacement in HHS is similar to that in DKA, although the fluid requirement may be greater due to the more extreme hyperosmolarity. By lowering plasma osmolality, IV fluid also improves insulin responsiveness and stress hormone levels. Sodium levels may need to be corrected in cases of extreme hyperosmolality. Insulin therapy starts similarly with an initial bolus of 0.1 to 0.2 U/kg and an infusion rate of approximately 0.1 to 0.2 U/kg/h. Instead of closing the anion gap, the goal is to bring the glucose into a reasonable range and then to normalize the osmolarity. While most patients with DKA are insulin sensitive, many patients with HHS are very insulin resistant. Much higher doses of insulin may be required. Once the BG falls to less than 300 mg/dL, the insulin infusion rate should be reduced by 50%. The serum glucose should be maintained between 250 and 300 mg/dL by adjusting the insulin infusion rate until the plasma osmolarity is less than 315 mOsm/L. Potassium repletion is similar to that in DKA with ½ NS being used for fluid repletion with 20 to 40 mEq/L of supplemental potassium.
V. NORMAL ADRENAL PHYSIOLOGY AND PATHOPHYSIOLOGY OF ADRENAL INSUFfiCIENCY
A. Adrenal Functional Anatomy. Each adrenal gland is made up of a cortex, which produces sex steroids, such as aldosterone and glucocorticoids, and a medulla, which produces adrenergic hormones such as epinephrine. The term “adrenal insufficiency” is commonly used to describe deficiency of both cortisol (which may be isolated) and aldosterone (which is almost always associated with cortisol deficiency).
B. Regulation of Adrenal Hormone Production (Fig. 27.2). Cortisol production by the adrenal glands is dependent on adrenocorticotrophic hormone (ACTH), which is produced by the pituitary gland. ACTH is regulated by corticotropin-releasing hormone (CRH), which is produced in the hypothalamus. Cortisol feeds back to inhibit CRH and ACTH release, closing the control loop. Cortisol deficiency may be caused by injury either to the adrenal cortex (primary adrenal insufficiency with elevated ACTH) or to the pituitary or hypothalamus (secondary or central adrenal insufficiency with low or “inappropriately normal” ACTH). Primary adrenal insufficiency is often associated with aldosterone deficiency, but central forms of adrenal insufficiency are limited to a deficit in cortisol production because aldosterone production is not dependent on ACTH.
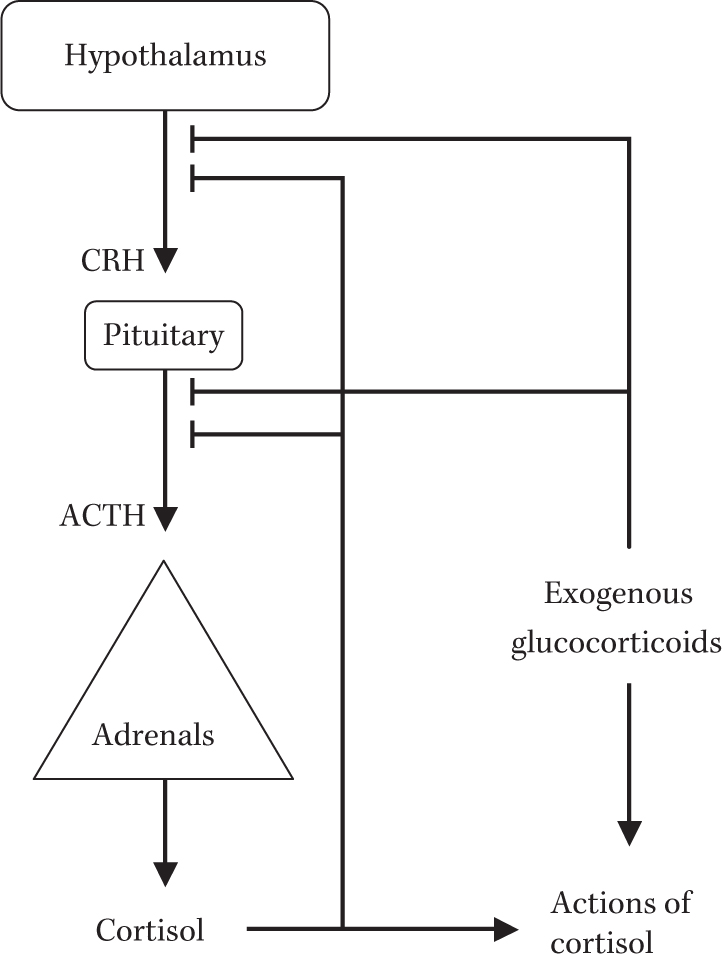
FIGURE 27.2 Regulation of adrenal hormone secretion. Arrows indicate positive action, production, or conversion. Lines ending in cross-bars indicate inhibition.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


