Cardiovascular Function and Vascular Tone: Physiology for ECC
Raúl J. Gazmuri
Beatrice M. Correa
When using potent vasoactive drugs in emergency cardiovascular care (ECC), specific hemodynamic goals should be established. These goals are aimed at preventing or reversing life-threatening conditions pending the reestablishment of effective hemodynamic function through spontaneous recovery or therapeutic intervention. Likewise, ECC providers should avoid prolonged use of these drugs without establishing the underlying hemodynamic diagnosis and instituting specific etiologic interventions.
During the treatment of cardiac arrest, pharmacologic support should be initiated as soon as possible after the rhythm is checked.
The required drugs should be anticipated and the agents prepared before a rhythm check so that they can be administered as soon as possible after the rhythm check.
Epinephrine 1 mg remains the initial recommended intravenous (IV) dose for shock-refractory ventricular fibrillation (VF). Higher additional doses may be harmful because of its beta-adrenergic effects.
Vasopressin has not been shown to elicit effects substantially different from those of epinephrine for the purpose of cardiac resuscitation. One dose of vasopressin 40 U may replace either the first or second dose of epinephrine in the treatment of cardiac arrest.
Overview: Pharmacology of Cardiac Function and Vascular Tone
In this chapter, drugs that can alter cardiac function and vascular tone are discussed in relation to their role in the management of cardiovascular emergencies, with primary focus on (1) hemodynamic stabilization to avert cardiac arrest, (2) resuscitation if cardiac arrest has occurred, and (3) management of postresuscitation hemodynamic dysfunction and related acute cardiovascular crises. From a didactic perspective, these drugs can be grouped into those that alter cardiac function and those that alter vascular tone. However, most of them affect cardiac function and vascular tone simultaneously in a dose-dependent manner, yielding complex physiologic effects compounded by potential side effects. An understanding of the cardiovascular effects of these drugs is of paramount importance for their proper use by ECC providers and in the proper clinical context.
Specific hemodynamic goals should be established in using these drugs, which are aimed at averting or reversing life-threatening conditions pending the reestablishment of competent hemodynamic function through spontaneous recovery or therapeutic intervention. Likewise, ECC providers should avoid prolonged use of these drugs without establishing the underlying hemodynamic diagnosis and instituting specific etiologic interventions. For example, use of vasopressor agents to reverse life-threatening hypotension associated with cardiac tamponade, massive pulmonary embolism, exsanguinating hemorrhage, or severe sepsis is appropriate, but only as a temporary measure pending definitive treatment.
Drugs targeting the cardiovascular system are used to (1) enhance myocardial contractility, (2) secure a physiologically appropriate rate and rhythm, and (3) increase or decrease vascular tone. This array of effects permits ECC providers to manipulate cardiac output and peripheral vascular resistance. However, these effects may come at the expense of unwanted side effects. Increasing contractility and heart rate will increase myocardial energy requirements, which could be detrimental, predisposing to myocardial ischemia when there is limited coronary blood supply (i.e., acute or chronic coronary artery obstruction compounded by hypotension). Inotropic agents also predispose to supraventricular and ventricular arrhythmias. Use of vasopressor agents will redistribute blood flow away from tissue beds that are not immediately vital (e.g., splanchnic territory, kidneys, skeletal muscle, skin), potentially leading to subsequent organ dysfunction.
Among the various drugs that affect the cardiovascular system, those that act through modulation of adrenergic receptors occupy a central role in the management of acute cardiovascular emergencies. A synopsis of adrenergic receptors and their physiology is, therefore, useful.
Adrenergic Receptors
Adrenergic receptors (adrenoceptors) are molecular structures present in plasma membranes. They all have seven transmembrane-spanning domains and are coupled to guanosine triphosphate–binding proteins (G proteins). After binding by specific extracellular ligands, they signal their biological effects through various cellular effector systems (Fig. 26-1). In the heart, adrenoceptors mediate changes in contractile function, pacemaker activity, and conduction velocity, leading to increases in cardiac output. In veins and arteries, adrenoceptors mediate changes in the tone of smooth muscle cells, prompting adaptive redistribution of blood flow through vasoconstriction or vasodilation of specific vascular beds. Adrenoceptors also mediate metabolic, endocrine, respiratory, gastrointestinal, urinary, and central nervous system function, but discussion of these effects exceeds the scope of this chapter. Adrenergic agonists include the endogenous catecholamines epinephrine and norepinephrine and sympathomimetic drugs such as isoproterenol, dobutamine, phenylephrine, and methoxamine (Table 26-1).
Two main adrenoceptors were originally described by Ahlquist1 based on the vascular response to endogenous agonists. Adrenoceptors that mediated vasoconstriction were designated as alpha adrenoceptors and those that mediated vasodilation as beta adrenoceptors. Subsequently it was discovered that these primary adrenoceptors included multiple subtypes, totaling, at the time of this writing, nine distinctive adrenoceptors (Fig. 26-2).2
Alpha Adrenoceptors
The main cardiovascular effect mediated by alpha adrenoceptor is vasoconstriction. This effect leads to redistribution of blood flow away from nonvital territories, favoring myocardial and cerebral perfusion under low-flow conditions. Alpha1 adrenoceptors also mediate inotropic effects, but with less potency than beta adrenoceptors. Two types
of alpha adrenoceptors have been identified: alpha1 and alpha2.
of alpha adrenoceptors have been identified: alpha1 and alpha2.
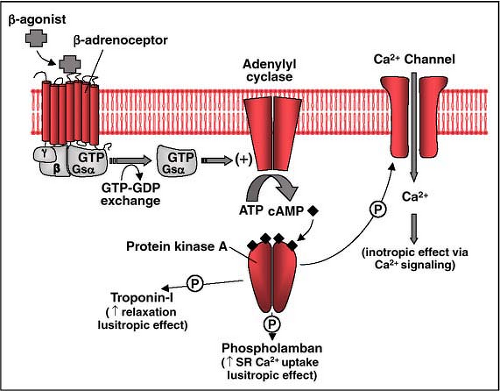 Figure 26-1 • Schematic representation of signal transduction through adrenergic receptors. Shown is a beta1 adrenoceptor with its seven transmembrane-spanning domains. The receptor is coupled to a heterotrimeric G protein signaling activation of adenylyl cyclase upon binding by its specific beta agonist (e.g., norepinephrine, epinephrine, isoproterenol, dobutamine). Adenylyl cyclase is activated by the stimulatory subunit Gsα and catalyzes the formation of cAMP from ATP. cAMP signals downstream activation of protein kinase A, which in turn phosphorylates Ca2+ channels, phospholamban, and troponin I, among other targets, enhancing contractile activity. GTP, guanosine triphosphate; GDT, guanosine triphosphate; alpha, beta, and gamma refer to subunits of the heterotrimeric GTP-binding protein; ATP, adenosine triphosphate; P, phosphate group; SR, sarcoplasmic reticulum. |
Table 26-1 • Drugs for Cardiovascular Support | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Alpha1 Adrenoceptors
These adrenoceptors are located postsynaptically in the immediate vicinity of sympathetic nerve terminals and in peripherally targeted organs.3 Alpha1 adrenoceptors signal their effects through Gq/11 and are coupled to several effector mechanisms, with the main mechanism linked to activation of phospholipase CB and inositol trisphosphate (IP3), causing increases in cytosolic Ca2+.4 In smooth muscle cells, Ca2+ activates Ca2+-sensitive kinases such as calmodulin-dependent myosin light-chain kinase, promoting contraction of smooth muscle cells (i.e., vasoconstriction). Three subtypes of alpha1 adrenoceptors have been
identified: alpha1A, alpha1B, and alpha1D. They have different localizations and activation patterns. Subtype alpha1A mediates vasoconstriction of specific arteries, including mammary, mesenteric, splenic, hepatic, omental, renal, pulmonary, and epicardial coronary, as well as specific veins, including the vena cava, saphenous, and pulmonary veins. Subtype alpha1D mediates contraction of the aorta and vena cava. In coronary arteries,5 alpha1-adrenoceptor stimulation may cause vasoconstriction if unopposed by beta2-adrenergic stimulation. In the myocardium, alpha1-adrenoceptors mediate a modest inotropic effect by increases in cytosolic Ca2+.6
identified: alpha1A, alpha1B, and alpha1D. They have different localizations and activation patterns. Subtype alpha1A mediates vasoconstriction of specific arteries, including mammary, mesenteric, splenic, hepatic, omental, renal, pulmonary, and epicardial coronary, as well as specific veins, including the vena cava, saphenous, and pulmonary veins. Subtype alpha1D mediates contraction of the aorta and vena cava. In coronary arteries,5 alpha1-adrenoceptor stimulation may cause vasoconstriction if unopposed by beta2-adrenergic stimulation. In the myocardium, alpha1-adrenoceptors mediate a modest inotropic effect by increases in cytosolic Ca2+.6
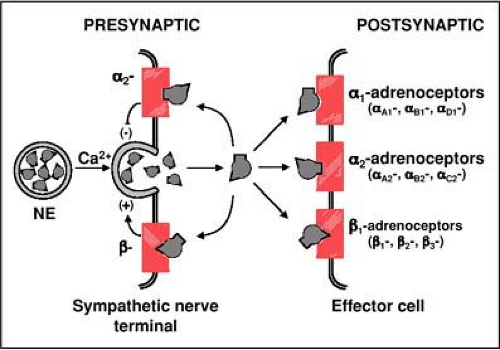 Figure 26-2 • Schematic representation of the various adrenoceptors, illustrating their presynaptic and postsynaptic locations. Note that agonists such as norepinephrine (NE) can activate alpha and beta adrenoceptors with different affinities, as illustrated in the figure by different “lock–key” configurations. In total there are three alpha1-adrenoceptors subtypes (alpha1A, alpha1B, and alpha1D), three alpha2-adrenoceptors subtypes (alpha2A, alpha2B, and alpha2C), and three beta-adrenoceptors types (beta1, beta2, and beta3). Alpha2 and beta adrenoceptors are also located presynaptically and modulate norepinephrine release. (Adapted from Tsuru H, Tanimitsu N, Hirai T. Role of perivascular sympathetic nerves and regional differences in the features of sympathetic innervation of the vascular system. Jpn J Pharmacol 2002;88:9–13, with permission.) |
Alpha2 Adrenoceptors
These adrenoceptors are located in the central nervous system, peripheral sympathetic nerve endings, and target organs. They are found presynaptically and postsynaptically.7 Three subtypes have been identified; alpha2A, alpha2B, and alpha2C. Presynaptic alpha2-adrenoceptors are predominantly alpha2A and alpha2C in the heart and all three in the adrenal gland.8,9 Their activation inhibits norepinephrine release (Fig. 26-2). Postsynaptic alpha2 adrenoceptors, centrally located, also reduce sympathetic flow such that central activation of presynaptic and postsynaptic alpha2 adrenoceptors causes a prominent and diffuse sympathoinhibitory response (i.e., explaining the antihypertensive effects of clonidine). Postsynaptic alpha2 adrenoceptors, located peripherally, are predominantly alpha2B and mediate contraction of smooth muscle cells, leading to vasoconstriction. These adrenoceptors (alpha2A) are also located distant from nerve endings in endothelial and smooth muscle cells and mediate vasoconstriction in response to circulating catecholamines.10 Experimentally, use of the alpha2-adrenoceptor agonist alpha methylnorepinephrine, which does not cross the blood–brain barrier, elicits a prominent vasoconstrictive response and attenuates cardiac sympathetic outflow. Both effects are desirable for resuscitation from cardiac arrest and in animal models improves resuscitation outcomes.11 This agent, however, is not available for clinical use.
Beta Adrenoceptors
The main cardiovascular effect of beta adrenoceptors is stimulation of cardiac activity and relaxation of vascular smooth muscle cells. Beta adrenoceptors mediate their effects through Gs, which activates adenylyl cyclase, enhancing the generation of cAMP, and in turn, signaling downstream through protein kinase A the phosphorylation of several kinases that regulate contractile activity.12 The same signaling mechanism enhances voltage-dependent Ca2+ channel activity (Fig. 26-1). Three types of beta adrenoceptors have been identified; beta1, beta2, and beta3. Beta1 and beta2 mediate cardiovascular responses. Beta3 has a lesser role in cardiovascular physiology but a prominent one in metabolism-mediating activity in white and brown adipose tissue.
Beta1 Adrenoceptors
These adrenoceptors are expressed predominantly in the heart, attaining high density in the sinus node and ventricular tissue. Their stimulation leads to increased myocardial contractility and relaxation, enhanced pacemaker activity, and increased conduction velocity, thus increasing overall cardiac activity as part of the stress response that follows sympathetic activation. Beta1 adrenoceptors are located postsynaptically, mostly in the immediate vicinity of sympathetic nerve terminals, but some are distributed more peripherally in target organs, to be stimulated by circulating catecholamines.
Beta2 Adrenoceptors
These adrenoceptors are present in smooth muscle cells of blood vessels. Their stimulation induces vasorelaxation also through signaling cAMP increase. They oppose the vasoconstrictive effects of alpha-adrenoceptor stimulation. Beta2 adrenoceptors are also present in the heart but with a different distribution than beta1 adrenoceptors, corresponding to approximately 20% of all beta adrenoceptors in left ventricular tissue but close to 50% in the sinus node, explaining the more prominent chronotropic effect of epinephrine and isoproterenol compared to norepinephrine, which lacks significant beta2-adrenergic agonist activity.3
Adrenergic Agonists
Adrenergic agonists include the endogenous catecholamines epinephrine and norepinephrine (Fig. 26-3). Epinephrine is
released from the adrenal glands, whereas norepinephrine is released from sympathetic postganglionic fibers. Epinephrine activates alpha1 and alpha2 and beta1 and beta2 adrenoceptors. Norepinephrine also activates alpha1, alpha2, and beta1 adrenoceptors but exerts minimal effects on beta2 adrenoceptors. Synthetic amines known as sympathomimetic drugs stimulate adrenoceptors, albeit with different adrenoceptor affinities and specificities, as shown in Table 26-1.
released from the adrenal glands, whereas norepinephrine is released from sympathetic postganglionic fibers. Epinephrine activates alpha1 and alpha2 and beta1 and beta2 adrenoceptors. Norepinephrine also activates alpha1, alpha2, and beta1 adrenoceptors but exerts minimal effects on beta2 adrenoceptors. Synthetic amines known as sympathomimetic drugs stimulate adrenoceptors, albeit with different adrenoceptor affinities and specificities, as shown in Table 26-1.
 Figure 26-3 • Metabolic steps in the synthesis of dopamine, norepinephrine, and epinephrine, with the involved enzymes shown in red. Note that dopamine is the immediate precursor of norepinephrine and epinephrine. Synthesis of norepinephrine is the final step in adrenergic sympathetic neurons. The subsequent step leading to epinephrine occurs in the adrenal medulla and in a few epinephrine-containing neuronal pathways in the brainstem. (Adapted from Nagatsu T. Genes for human catecholamine-synthesizing enzymes. Neurosci Res 1991;12:315–345, with permission.) |
Dopamine Receptors
Dopamine (D) receptors are also G protein–coupled receptors. To date, five types have been identified and grouped into two main types based on whether they are coupled to stimulatory (Gsα) or inhibitory (Giα) G-proteins. Thus, D1 receptors (including D1 and D5) are coupled to Gsα and stimulate formation of cAMP by increasing adenylyl cyclase activity. D2 receptors (including D2, D3, and D4) are coupled to Giα and have the opposite effect reducing the formation of cAMP by inhibiting adenylyl cyclase. D receptors play an important role in neurologic processes such as the control of motivation, learning, and fine motor movement.13 They also mediate neuroendocrine signaling by modulating the release of prolactin and thyroid hormone from the pituitary gland. Abnormalities in D receptors and dopaminergic nerve function have been implicated in several neuropsychiatric disorders, such as Parkinson’s disease, drug addiction, compulsive behavior, attention-deficit/hyperactivity disorder, and schizophrenia.13
In the cardiovascular system, D receptors mediate selected vasodilatory responses along with inotropic and chronotropic cardiac responses. In the kidneys, D receptors mediate increased glomerular filtration, diuresis, and natriuresis.14 These cardiovascular and renal effects are mediated through D1 receptors located postsynaptically in vascular smooth muscle cells and kidney. D2 receptors are located presynaptically at various sites within the sympathetic nervous system. Their activation leads to inhibition of sympathetic nervous system activity.
Dopamine
Dopamine is the naturally occurring agonist of D receptors. Given exogenously at low doses, it promotes vasodilation and increased renal function with minimal central effect because it does not readily cross the blood–brain barrier. Main target vascular beds include cerebral, renal, splanchnic, and coronary arteries. At somewhat higher doses, dopamine exerts inotropic effect by stimulating beta1 adrenoceptors; at even higher doses, it enhances norepinephrine synthesis and release from sympathetic nerve terminals, leading to vasoconstriction, because it is the immediate metabolic precursor in the synthesis of norepinephrine (Fig. 26-3).15
Integrated Adrenergic Response
The broad diversity of adrenoceptors presented above permits, at the cellular level, signaling of distinct responses contingent on cell type, effector systems, and target mechanisms. At the organ level, receptor density, specific subtypes, and relative ligand potency allow for specific responses to sympathetic stimulation, circulating catecholamines, and sympathomimetic agents. For example, beta1 adrenoceptors—as discussed above—are distributed predominantly in cardiac tissue, mediating inotropic and chronotropic responses. Blood vessels express alpha and beta adrenoceptors in varying proportions and densities, leading to vasoconstrictive or vasodilatory responses after stimulation by endogenous catecholamines or sympathomimetic drugs contingent on the vascular bed and local concentration of the agonists. For example, cutaneous vessels express almost exclusively alpha adrenoceptors, such that stimulation by epinephrine or norepinephrine leads to vasoconstriction; whereas stimulation by isoproterenol has virtually no effects. Blood vessels that supply skeletal muscles express beta2 and alpha adrenoceptors, such that the predominant effect becomes contingent on the dose of the adrenergic agent. Epinephrine at low doses causes vasodilation, reflecting the predominance of beta2-agonist effect, given a higher receptor affinity, whereas at higher doses, the same epinephrine causes vasoconstriction as the
alpha-adrenoceptor effect predominates. This pharmacologic effect is useful during cardiac resuscitation when exogenous epinephrine is given to redistribute blood toward the heart and brain. Dopamine, the natural agonist of dopamine receptor and precursor of norepinephrine and epinephrine, mediates inotropic and vasodilatory responses at relatively low doses. However, as the dopamine dose increases, vasoconstrictive responses occur, related to increased production of norepinephrine in sympathetic nerve terminals.
alpha-adrenoceptor effect predominates. This pharmacologic effect is useful during cardiac resuscitation when exogenous epinephrine is given to redistribute blood toward the heart and brain. Dopamine, the natural agonist of dopamine receptor and precursor of norepinephrine and epinephrine, mediates inotropic and vasodilatory responses at relatively low doses. However, as the dopamine dose increases, vasoconstrictive responses occur, related to increased production of norepinephrine in sympathetic nerve terminals.
Besides the primary effect of adrenergic stimulation, many additional factors influence the clinical response to adrenergic agonists, including the baroreflex response slowing the heart rate, activity of the parasympathetic nervous system, release of cotransmitters including ATP and neuropeptide Y from sympathetic nerve terminals, the effects of vasoactive platelet–mediated products such as thromboxane A2 and prostacyclin, endothelial injury causing paradoxical responses to vasodilating stimuli, and interactions with other drugs.
Accordingly, the clinical effects of adrenergic agonists may vary widely from patient to patient and is further influenced by disease states, including whether the patient is in cardiac arrest. Because of this variability, these drugs may need to be titrated and the patient response closely monitored. In Table 26-1, a simplified selection of vasoactive drugs is summarized, identifying their major receptor sites and their effects on arterial vascular tone, heart rate, myocardial contractility, and the risk for inducing arrhythmias. Their usual clinical doses are also given.
Vasopressor Agents Used for Cardiac Resuscitation
Cessation of coronary blood flow after cardiac arrest leads to rapid development of intense global myocardial ischemia, especially when VF is the precipitating mechanism.16 Unless spontaneous cardiac activity is promptly restored, as when VF is terminated by immediate delivery of electrical shocks (i.e., within a few minutes),17 successful resuscitation relies on providers’ capability for generating coronary blood flow above critical threshold levels.18 Such blood flow is proportional to the pressure gradient established between the aorta and the right atrium during the relaxation phase of chest compression (i.e., coronary perfusion pressure).19,20 Critical threshold levels predictive of resuscitation have been identified in various animal models and in human victims of cardiac arrest, corresponding to 20 mm Hg in rats21 and dogs,22 10 mm Hg in pigs,23 and 15 mm Hg in humans. Once the resuscitability threshold is exceeded, proportionally greater resuscitability rates are expected18 (Fig. 26-4).
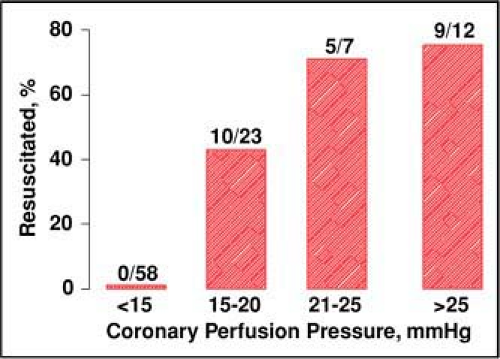 Figure 26-4 • Relationship between coronary perfusion pressure and initial resuscitation rates (%) in 100 victims of out-of-hospital cardiac arrest. Victims who failed initial resuscitation attempts at the scene were brought the emergency department and instrumented to measure coronary perfusion pressure during ongoing chest compression. A minimum coronary perfusion pressure of 15 mm Hg was noted before return of spontaneous circulation could be reestablished. Resuscitation rates increased proportional to coronary perfusion pressure after such threshold was exceeded. (Adapted from Paradis NA, Martin GB, Rivers EP, et al. Coronary perfusion pressure and the return of spontaneous circulation in human cardiopulmonary resuscitation. JAMA 1990;263:1106–1113, with permission.) |
To increase the coronary perfusion pressure, providers must pay close attention to the technique for generating forward blood flow. Current guidelines emphasize the importance of ensuring optimal depth and rate of compression, full chest recoil between compressions, avoiding hyperventilation, and minimizing interruptions of chest compression.24 However, the forward blood flow generated by chest compression rarely exceeds 20% of the normal cardiac output20,25 and typically deteriorates over time.26 Therefore, increases in systemic vascular resistance are required to increase the coronary perfusion pressure above resuscitability thresholds. Although a prominent neuroendocrine stress response, triggered by cardiac arrest, prompts peripheral vasoconstriction, its magnitude is limited and exogenous administration of vasoconstrictive agents is often necessary.
The 2005 guidelines recognized epinephrine and vasopressin as the preferred vasopressor agents for cardiac resuscitation.24 Yet to date, no placebo-controlled trial has been conducted in humans to determine whether vasopressor agents ultimately increase the rate of neurologically intact survival to hospital discharge.
Epinephrine
Epinephrine is hemodynamically beneficial during cardiac resuscitation because of its alpha-adrenoceptor vasoconstrictive
effect.27 In animal models, alpha-adrenergic stimulation during cardiac resuscitation increases coronary and cerebral blood flow and favors return of spontaneous circulation.28 Experimentally, differences in the effects mediated through alpha1 and alpha2 adrenoceptors appear relevant. Alpha1 adrenoceptors desensitize faster than alpha2 adrenoceptors during ischemia and acidosis, such that there is a better alpha2-mediated vasoconstrictive response after prolonged cardiac arrest. Alpha1 adrenoceptors signal a modest inotropic effect through increases in cytosolic Ca2+, which can be detrimental by increasing myocardial energy requirements and inducing arrhythmias. In contrast, presynaptic alpha2 adrenoceptors in the heart limit the release of norepinephrine, exerting a favorable antiadrenergic effect.8 Concomitant beta-adrenergic stimulation by epinephrine may be detrimental by increasing myocardial energy demands. Animal studies have shown that marked increases in myocardial blood flow by epinephrine during chest compression fail to attenuate ischemia, such that intramyocardial lactate and ATP depletion remain unchanged (Fig. 26-5).29 In other studies, epinephrine has been shown to worsen postresuscitation myocardial function. Such detrimental effects can be reduced by concomitant use of beta-blockers or by use of selective alpha agonists instead of epinephrine.30
effect.27 In animal models, alpha-adrenergic stimulation during cardiac resuscitation increases coronary and cerebral blood flow and favors return of spontaneous circulation.28 Experimentally, differences in the effects mediated through alpha1 and alpha2 adrenoceptors appear relevant. Alpha1 adrenoceptors desensitize faster than alpha2 adrenoceptors during ischemia and acidosis, such that there is a better alpha2-mediated vasoconstrictive response after prolonged cardiac arrest. Alpha1 adrenoceptors signal a modest inotropic effect through increases in cytosolic Ca2+, which can be detrimental by increasing myocardial energy requirements and inducing arrhythmias. In contrast, presynaptic alpha2 adrenoceptors in the heart limit the release of norepinephrine, exerting a favorable antiadrenergic effect.8 Concomitant beta-adrenergic stimulation by epinephrine may be detrimental by increasing myocardial energy demands. Animal studies have shown that marked increases in myocardial blood flow by epinephrine during chest compression fail to attenuate ischemia, such that intramyocardial lactate and ATP depletion remain unchanged (Fig. 26-5).29 In other studies, epinephrine has been shown to worsen postresuscitation myocardial function. Such detrimental effects can be reduced by concomitant use of beta-blockers or by use of selective alpha agonists instead of epinephrine.30
Despite these considerations, epinephrine is the most used vasopressor agent for resuscitation from cardiac arrest. Previous investigators had suggested that higher initial doses or escalating doses could improve survival rates. However, eight randomized clinical studies—involving more than 9,000 cardiac arrest victims—failed to demonstrate that higher doses of epinephrine improve survival to hospital discharge or neurologic outcomes when compared with standard doses.31,32,33,34,35,
 Emergency Department Management of Acute Coronary Syndromes
Emergency Department Management of Acute Coronary Syndromes
 Heart Failure and Acute Pulmonary Edema in the Emergency Department
Heart Failure and Acute Pulmonary Edema in the Emergency Department
 Circulatory Adjuncts to Improve Coronary Perfusion Pressure
Circulatory Adjuncts to Improve Coronary Perfusion Pressure
 Basic Life Support: Science to Survival
Basic Life Support: Science to Survival
 Endotracheal Intubation and Management of the Difficult Airway
Endotracheal Intubation and Management of the Difficult Airway
 Bradycardia
Bradycardia




Related posts:
Emergency Department Management of Acute Coronary Syndromes
Heart Failure and Acute Pulmonary Edema in the Emergency Department
Circulatory Adjuncts to Improve Coronary Perfusion Pressure
Endotracheal Intubation and Management of the Difficult Airway
Bradycardia
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
