been thoroughly defined and described by Hoffman, are summarized in the following text and in Figure 36.2.2
TABLE 36.1 Factor Nomenclature and Half-Lives | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||
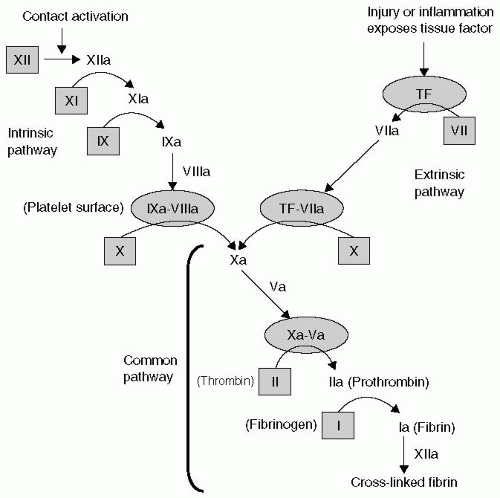 FIGURE 36.1 The intrinsic and extrinsic pathways of coagulation. Factors in the inactive form are within shaded squares. The shaded ovals represent phospholipid surfaces (tissue factor [TF] or platelets). |
prothrombinase complex supports the amplification of the coagulation process in four ways, which are as follows:
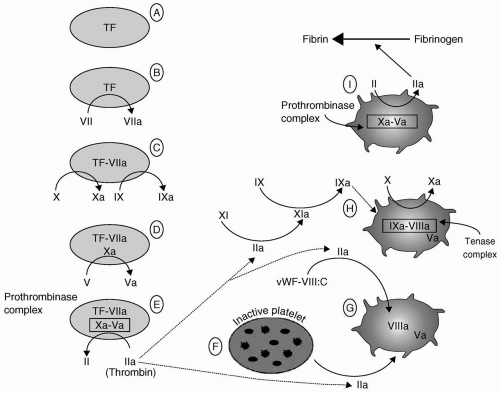 FIGURE 36.2 The coagulation mechanism. See text for a detailed explanation. TF, extravascular membrane-bound tissue factor; vWF-VIII:C, circulating factor VIII bound to its carrier protein, the von Willebrand factor. (Reproduced with permission from Drummond JC, Petrovitch CT. Hemotherapy and hemostasis. In: Barash PG, Cullen BF, Stoelting RK, eds. Clinical anesthesia, 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2006:208.) |
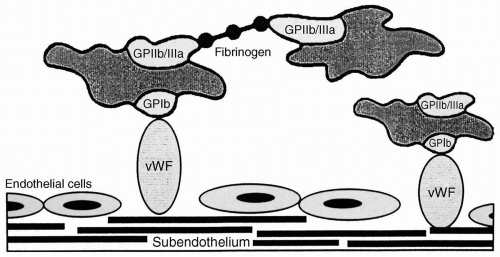 FIGURE 36.3 Platelet adhesion and aggregation. When the endothelium is denuded, von Willebrand factor (vWF) binds to collagen in the subendothelial layer. Platelets adhere through their glycoprotein 1b (GPIb)-IX-V receptors to vWF. Platelets aggregate to one another by cross-linking through fibrinogen (or vWF, not shown) between GPIIb-IIIa receptors expressed on the platelet surface during the process of platelet activation. (Reproduced with permission from Drummond JC, Petrovitch CT. Hemotherapy and hemostasis. In: Barash PG, Cullen BF, Stoelting RK, eds. Clinical anesthesia, 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2006:208.) |
First, it further activates the adjacent platelets (Fig. 36.2F). That activation results in platelet surface changes, most notably the appearance of the GPIIb-IIIa receptor, and the release of the contents of platelet granules. Among those contents are adenosine diphosphate (ADP), a powerful platelet activator and proaggregant that rapidly recruits other platelets to the growing platelet mass, and FV.
Thrombin’s second effect promotes the activation of FV to FVa (Fig. 36.2G).
Third, thrombin releases circulating FVIII from its carrier molecule (vWF) and activates it (Fig. 36.2G).
Fourth, thrombin activates factor XI (see Fig. 36.2H). Factor XIa in turn activates factor IX (Fig. 36.2H), further adding to the pool of factor IXa that first formed during the activation phase above (Fig. 36.2C). The net result of this amplification stage is the availability of activated platelets and activated factors V, VIII, and IX.
Type I (70% to 80% of vWD) is a quantitative defect. A functional vWF protein is secreted in reduced amount. Type I vWD presents with the mucocutaneous bleeding that is characteristic of abnormalities of primary hemostasis, that is, appears clinically as a platelet defect.
Type II vWD (20% to 30% of vWD) includes numerous qualitative defects of vWF. Some mutations affect the vWF’s interaction with platelets and others affect the interaction with factor VIII. Type II is subdivided into four subtypes. IIB is characterized by vWF molecule that causes abnormal aggregation of platelets and thrombocytopenia. The abnormal vWF in type IIB has a high affinity for the platelet GPIb receptor. The bleeding diathesis is probably the result of formation and clearance of vWF-platelet complexes and the resultant thrombocytopenia. 1-Deamino-8-D-arginine vasopressin (DDAVP), which promotes the release of vWF from the endothelium, will aggravate this variant of vWD. In the subtype IIN (Normandy), the vWF has a markedly reduced affinity for factor VIII. These patients demonstrate normal platelet function, but bleed because of decreased factor VIII coagulant activity. These patients are readily misdiagnosed as having mild hemophilia A.
Type III (very rare) is characterized by the complete absence of vWF, resulting in a severe abnormality of both primary hemostasis and coagulation.
that the results of common coagulation tests (platelet count, aPTT, PT) may be normal in the patient with vWD. Although the half-life of VIII:C is diminished in vWD, there is usually sufficient factor VIII:C to yield a normal aPTT in basal conditions.
that is clinically indistinguishable from hemophilia A. Typically, minor spontaneous hemorrhage is managed by achieving factor IX levels of 20% to 30% of normal. Levels of 50% to 100% are targeted in the event of more severe hemorrhage and in anticipation of elective surgery. Factor IX complex concentrates, also known as prothrombin complex concentrates (II, VII, IX, X), have been used in the face of resistance to factor IX concentrates. However, they convey an infectious hazard and may entail the risk of thrombosis and DIC because of the presence of activated factors. An rFIX concentrate is now available and is the preferred therapy.
TABLE 36.2 Causes of Thrombocytopenia | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
are unable to adhere to phospholipid surfaces during the coagulation process. When vitamin K deficiency occurs, the K-dependent factors are depleted in an order determined by their half-lives. Factor VII has the shortest half-life and is depleted first, followed by factors IX and X, and finally factor II.
LOW MOLECULAR WEIGHT HEPARIN (LMWH): Low molecular weight fractions of heparin are supplanting subcutaneous unfractionated heparin and coumadin for DVT prophylaxis and treatment.31 There are several available agents, including certoparin, dalteparin, danaparoid, enoxaparin, reviparin, and tinzaparin. These agents do not appear to differ in terms of efficacy.32 Enoxaparin is used most widely in the United States. LMWHs also act through ATIII but have greater activity against factor Xa than thrombin (IIa). The ratio of Xa/IIa activity varies among the agents (enoxaparin 3.8:1; tinzaparin 1.9:1).33 As a consequence, the effect of these agents on standard coagulation tests will vary (minimal for enoxaparin34), as will the effect of protamine neutralization, which is very incomplete for enoxaparin. Coagulation testing is usually not required or performed. If laboratory testing is deemed necessary, the anti-Xa level is the appropriate test. LMWH causes less platelet inhibition and is associated with a lesser incidence of heparin-induced thrombocytopenia.35 There has been considerable variation in the dosage regimens employed. In Europe, it has been common to begin prophylactic administration 12 to 24 hours preoperatively. Postprocedure administration is more common in North America, but there is little evidence to suggest the superiority of either practice.32 Twice daily dosing with enoxaparin has been common in North America. However, once-daily regimens are usually sufficient and may actually be safer in some circumstances. Because of the relatively long half-life of enoxaparin, twice daily dosing poses a problem with
respect to removal of epidural catheters because there is no anticoagulant nadir.
HEPARIN-INDUCED THROMBOCYTOPENIA/THROMBOSIS (HITT): The clinical manifestations of heparin-induced thrombocytopenia/thrombosis (HITT) are most commonly thrombotic and thromboembolic events (DVT, PE, limb or acral ischemia, MI, stroke), rather than a bleeding diathesis.36 Accordingly, this chapter will not provide a detailed description. As many as 5% of patients who receive heparin therapy for 5 days will develop thrombocytopenia that results from the development of antibodies (usually immunoglobulin G [IgG]) directed against platelet factor 4-heparin complexes. HITT appears to be dose-related and is more common with bovine than porcine heparin. Onset usually occurs after several days in the heparin naive patient but can occur much more quickly (10 to 12 hours) in those exposed within the preceding 100 days.37 LMWH-associated HITT occurs at a much lesser frequency and requires longer periods of exposure.38 Treatment requires withdrawal of heparin and institution of an alternate anticoagulant (e.g., a direct thrombin inhibitor [DTI] such as hirudin, argatroban, lepirudin, bivalirudin or a heparinoid such as danaparoid), but not a LMWH. Warfarin is contraindicated because the inhibition of proteins C and S by warfarin in the face of ongoing platelet clumping may aggravate thrombosis. Platelets may similarly contribute to thrombosis and should not be administered unless thrombocytopenia is extreme.
HEPARIN IN CARDIOPULMONARY BYPASS: A comprehensive review of the use and monitoring of heparin therapy in cardiopulmonary bypass (CPB) is beyond the scope of this chapter, and extensive reviews are available elsewhere.39 In brief, the common practice is to administer sufficient heparin to maintain ACT >480 to 500 seconds for the duration of bypass. Although there is no universal agreement, it appears that there is greater hazard in allowing ACT to be on the “low side” than in maintaining more complete heparinization.40 Platelet activation is less apparent when longer ACTs are maintained. Whether this is a function of direct inhibition of platelets, which are subject to contact activation by the CPB circuit, binding of vWF or the result of reduced formation of thrombin and inhibition of platelets by fibrin breakdown products (FDPs) is not apparent to the authors of this chapter. Protamine is used to reverse heparin’s effect. Many clinicians employ a “milliliter for a milliliter” technique. However, titration of protamine against ACT is ideal to avoid the excessive administration of protamine, which has inherent anticoagulant effects including platelet inhibition, stimulation of tPA release from endothelium, and inhibition of fibrinogen cleavage by thrombin.41 Various alternatives have been used for the patient with HIT who requires CPB. Plasmapheresis before surgery with subsequent use of heparin has been reported.42 Nonheparin anticoagulants, including the defibrinogenating agent Ancrod (from the venom of the Malaysian pit viper) and, more commonly DTIs, have been employed.43 The contact activation of platelets is not inhibited, and it may be appropriate to administer platelet inhibitors simultaneously.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


