Key Concepts
 Weakness associated with myasthenia gravis is due to autoimmune destruction or inactivation of postsynaptic acetylcholine receptors at the neuromuscular junction, leading to reduced numbers of receptors and degradation of their function, and to complement-mediated damage to the postsynaptic membrane.
Weakness associated with myasthenia gravis is due to autoimmune destruction or inactivation of postsynaptic acetylcholine receptors at the neuromuscular junction, leading to reduced numbers of receptors and degradation of their function, and to complement-mediated damage to the postsynaptic membrane.
 Patients who have myasthenia gravis with respiratory muscle or bulbar involvement are at increased risk for pulmonary aspiration.
Patients who have myasthenia gravis with respiratory muscle or bulbar involvement are at increased risk for pulmonary aspiration.
 Many patients with myasthenia gravis are exquisitely sensitive to nondepolarizing neuromuscular blockers (NMBs).
Many patients with myasthenia gravis are exquisitely sensitive to nondepolarizing neuromuscular blockers (NMBs).
 Patients who have myasthenia gravis are at risk for postoperative respiratory failure. Disease duration of more than 6 years, concomitant pulmonary disease, a peak inspiratory pressure of less than −25 cm H2O (ie, −20 cm H2O), a vital capacity less than 4 mL/kg, and a pyridostigmine dose greater than 750 mg/d are predictive of the need for postoperative ventilation following thymectomy.
Patients who have myasthenia gravis are at risk for postoperative respiratory failure. Disease duration of more than 6 years, concomitant pulmonary disease, a peak inspiratory pressure of less than −25 cm H2O (ie, −20 cm H2O), a vital capacity less than 4 mL/kg, and a pyridostigmine dose greater than 750 mg/d are predictive of the need for postoperative ventilation following thymectomy.
 Patients with Lambert-Eaton myasthenic syndrome and other paraneoplastic neuromuscular syndromes are very sensitive to both depolarizing and nondepolarizing NMBs.
Patients with Lambert-Eaton myasthenic syndrome and other paraneoplastic neuromuscular syndromes are very sensitive to both depolarizing and nondepolarizing NMBs.
 Respiratory muscle degeneration in patients with muscular dystrophy interferes with an effective cough mechanism and leads to retention of secretions and frequent pulmonary infections.
Respiratory muscle degeneration in patients with muscular dystrophy interferes with an effective cough mechanism and leads to retention of secretions and frequent pulmonary infections.
 Degeneration of cardiac muscle in patients with muscular dystrophy is also common, but results in dilated or hypertrophic cardiomyopathy in only 10% of patients.
Degeneration of cardiac muscle in patients with muscular dystrophy is also common, but results in dilated or hypertrophic cardiomyopathy in only 10% of patients.
 Succinylcholine should be avoided in patients with Duchenne’s or Becker’s muscular dystrophies because of unpredictable response and the risk of inducing severe hyperkalemia or triggering malignant hyperthermia.
Succinylcholine should be avoided in patients with Duchenne’s or Becker’s muscular dystrophies because of unpredictable response and the risk of inducing severe hyperkalemia or triggering malignant hyperthermia.
 Anesthetic management in patients with periodic paralysis is directed toward preventing attacks. Intraoperative management should include frequent determinations of plasma potassium concentrations and careful electrocardiographic monitoring to detect arrhythmias.
Anesthetic management in patients with periodic paralysis is directed toward preventing attacks. Intraoperative management should include frequent determinations of plasma potassium concentrations and careful electrocardiographic monitoring to detect arrhythmias.
 In patients with periodic paralysis, the response to NMBs is unpredictable, and neuromuscular function should be carefully monitored during their use. Increased sensitivity to nondepolarizing NMBs is particularly apt to be encountered in patients with hypokalemic periodic paralysis.
In patients with periodic paralysis, the response to NMBs is unpredictable, and neuromuscular function should be carefully monitored during their use. Increased sensitivity to nondepolarizing NMBs is particularly apt to be encountered in patients with hypokalemic periodic paralysis.
Anesthesia for Patients with Neuromuscular Disease: Introduction
Although neuromuscular diseases are relatively uncommon, patients with these conditions will present to the operating room and to non-operating room procedure areas for diagnostic studies, treatment of complications, or surgical management of related or unrelated disorders. Overall debility, with diminished respiratory muscle strength and increased sensitivity to neuromuscular blockers (NMBs), predisposes these patients to postoperative ventilatory failure and pulmonary aspiration, and may slow their post-procedure recovery because of difficulty with ambulation and increased risk of falling. A basic understanding of the major disorders and their potential interaction with anesthetic agents is necessary to minimize the risk of perioperative morbidity.
Myasthenia Gravis
Myasthenia gravis is an autoimmune disorder characterized by weakness and easy fatigability of skeletal muscle. It is classified according to disease distribution and severity (Table 35-1). The prevalence is estimated at 50-200 per million population. The incidence is highest in women during their third decade, and men exhibit two peaks, one in the third decade and another in the sixth decade.
| Class | Definition |
|---|---|
| I |
|
| II |
|
| IIa |
|
| IIb |
|
| III |
|
| IIIa |
|
| IIIb |
|
| IV |
|
| IVa |
|
| IVb |
|
| V | Defined by intubation, with or without mechanical ventilation, except when employed during routine postoperative management. The use of a feeding tube without intubation places the patient in class IVb |
 Weakness associated with myasthenia gravis is due to autoimmune destruction or inactivation of postsynaptic acetylcholine receptors at the neuromuscular junction, leading to reduced numbers of receptors and degradation of their function, and to complement-mediated damage to the postsynaptic end plate. IgG antibodies against the nicotinic acetylcholine receptor in neuromuscular junctions are found in 85-90% of patients with generalized myasthenia gravis and up to 50-70% of patients with ocular myasthenia. Among patients with myasthenia, 10-15% percent develop thymoma, whereas approximately 70% exhibit histologic evidence of thymic lymphoid follicular hyperplasia. Other autoimmune-related disorders (hypothyroidism, hyperthyroidism, rheumatoid arthritis, and systemic lupus erythematosus) are also present in up to 10% of patients. The differential diagnosis of myasthenia gravis includes a number of other clinical conditions that may mimic its signs and symptoms (Table 35-2). Myasthenia gravis crisis is an exacerbation requiring mechanical ventilation and should be suspected in any patient with respiratory failure of unclear etiology.
Weakness associated with myasthenia gravis is due to autoimmune destruction or inactivation of postsynaptic acetylcholine receptors at the neuromuscular junction, leading to reduced numbers of receptors and degradation of their function, and to complement-mediated damage to the postsynaptic end plate. IgG antibodies against the nicotinic acetylcholine receptor in neuromuscular junctions are found in 85-90% of patients with generalized myasthenia gravis and up to 50-70% of patients with ocular myasthenia. Among patients with myasthenia, 10-15% percent develop thymoma, whereas approximately 70% exhibit histologic evidence of thymic lymphoid follicular hyperplasia. Other autoimmune-related disorders (hypothyroidism, hyperthyroidism, rheumatoid arthritis, and systemic lupus erythematosus) are also present in up to 10% of patients. The differential diagnosis of myasthenia gravis includes a number of other clinical conditions that may mimic its signs and symptoms (Table 35-2). Myasthenia gravis crisis is an exacerbation requiring mechanical ventilation and should be suspected in any patient with respiratory failure of unclear etiology.
Other neuromuscular disorders
|
Cranial nerve palsies
|
Muscle disease
|
Central nervous system pathology
|
Other
|
The course of myasthenia gravis is marked by exacerbations and remissions, which may be partial or complete. The weakness can be asymmetric, confined to one group of muscles, or generalized. Ocular muscles are most commonly affected, resulting in fluctuating ptosis and diplopia. With bulbar involvement, laryngeal and pharyngeal muscle weakness can result in dysarthria, difficulty in chewing and swallowing, problems clearing secretions, or pulmonary aspiration. Severe disease is usually also associated with proximal muscle weakness (primarily in the neck and shoulders) and involvement of respiratory muscles. Muscle strength characteristically improves with rest but deteriorates rapidly with exertion. Infection, stress, surgery, and pregnancy have unpredictable effects on the disease but often lead to exacerbations. A number of medications may exacerbate the signs and symptoms of myasthenia gravis (Table 35-3).
Cardiovascular agents
|
Antibiotics
|
Central nervous system drugs
|
Immunomodulators
|
Rheumatological agents
|
Miscellanous
|
Anticholinesterase drugs are used most commonly to treat the muscle weakness of this disorder. These drugs increase the amount of acetylcholine at the neuromuscular junction through inhibition of end plate acetylcholinesterase. Pyridostigmine is prescribed most often; when given orally, it has an effective duration of 2-4 h. Excessive administration of an anticholinesterase may precipitate cholinergic crisis, which is characterized by increased weakness and excessive muscarinic effects, including salivation, diarrhea, miosis, and bradycardia. An edrophonium (Tensilon) test may help differentiate a cholinergic from a myasthenic crisis. Increased weakness after administration of up to 10 mg of intravenous edrophonium indicates cholinergic crisis, whereas increasing strength implies myasthenic crisis. If this test is equivocal or if the patient clearly has manifestations of cholinergic hyperactivity, all cholinesterase drugs should be discontinued and the patient should be monitored in an intensive care unit or close-observation area. Anticholinesterase drugs are often the only agents used to treat patients with mild disease. Moderate to severe disease is treated with a combination of an anticholinesterase drug and immunomodulating therapy. Corticosteroids are usually tried first, followed by azathioprine, cyclosporine, cyclophosphamide, mycophenolate mofetil, and intravenous immunoglobulin. Plasmapheresis is reserved for patients with dysphagia or respiratory failure, or to normalize muscle strength preoperatively in patients undergoing a surgical procedure, including thymectomy. Up to 85% of patients younger than 55 years of age show clinical improvement following thymectomy even in the absence of a tumor, but improvement may be delayed up to several years.
Patients with myasthenia gravis may present for thymectomy or for unrelated surgical or obstetric procedures, and medical management of their condition should be optimized prior to the intended procedure. Myasthenic patients with respiratory and oropharyngeal weakness should be treated preoperatively with intravenous immunoglobulin or plasmapheresis. If strength normalizes, the incidence of postoperative respiratory complications should be similar to that of a nonmyasthenic patient undergoing a similar surgical procedure. Patients scheduled for thymectomy may have deteriorating muscle strength, whereas those undergoing other elective procedures may be well controlled or in remission. Adjustments in anticholinesterase medication, immunosuppressants, or steroid therapy in the perioperative period may be necessary. Patients with advanced generalized disease may deteriorate significantly when anticholinesterase agents are withheld. These medications should be restarted when the patient resumes oral intake postoperatively. When necessary, cholinesterase inhibitors can also be given parenterally at 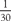 the oral dose. Potential problems associated with management of anticholinesterase therapy in the postoperative period include altered patient requirements, increased vagal reflexes, and the possibility of disrupting bowel anastomoses secondary to hyperperistalsis. Moreover, because these agents also inhibit plasma cholinesterase, they could theoretically prolong the duration of ester-type local anesthetics and succinylcholine.
the oral dose. Potential problems associated with management of anticholinesterase therapy in the postoperative period include altered patient requirements, increased vagal reflexes, and the possibility of disrupting bowel anastomoses secondary to hyperperistalsis. Moreover, because these agents also inhibit plasma cholinesterase, they could theoretically prolong the duration of ester-type local anesthetics and succinylcholine.
Preoperative evaluation should focus on the recent course of the disease, the muscle groups affected, drug therapy, and coexisting illnesses.  Patients who have myasthenia gravis with respiratory muscle or bulbar involvement are at increased risk for pulmonary aspiration. Premedication with metoclopramide or an H2 blocker or proton pump inhibitor may decrease this risk. Because patients with myasthenia are often very sensitive to the respiratory depressant effect of opioids and benzodiazepines, premedication with these drugs should be done with caution, if at all.
Patients who have myasthenia gravis with respiratory muscle or bulbar involvement are at increased risk for pulmonary aspiration. Premedication with metoclopramide or an H2 blocker or proton pump inhibitor may decrease this risk. Because patients with myasthenia are often very sensitive to the respiratory depressant effect of opioids and benzodiazepines, premedication with these drugs should be done with caution, if at all.
With the exception of NMBs, standard anesthetic agents may be used in patients with myasthenia gravis. Marked respiratory depression, however, may be encountered following even moderate doses of propofol or opioids. When general anesthesia is required, a volatile agent-based anesthetic is frequently employed. Deep anesthesia with a volatile agent alone in patients with myasthenia may provide sufficient relaxation for tracheal intubation and most surgical procedures, and many clinicians routinely avoid NMBs entirely. The response to succinylcholine is said to be unpredictable, but we have not found this to be so in practice. Patients may manifest a relative resistance, or a moderately prolonged effect (see Chapter 11). The dose of succinylcholine may be increased to 2 mg/kg to overcome any resistance, expecting that the duration of paralysis could be increased by 5-10 min.





