ATN results from a variety of synergistic combinations of ischemic and nephrotoxic insults rather than from any single insult.
1,
24 It is characterized by a complex interaction between the injured tubular epithelial cells and the vascular smooth muscle and endothelial cells, most likely genetically determined by the capacity of each group of these cells to regenerate and recover.
25,
26,
27 Pioneering studies of ATN pathophysiology in the early 1940s indicated a uniform pattern of disturbed renal function, which was characterized by gross tubular dysfunction and extreme diminution in renal blood flow (RBF), coupled with anatomic evidence of tubular necrosis and regeneration.
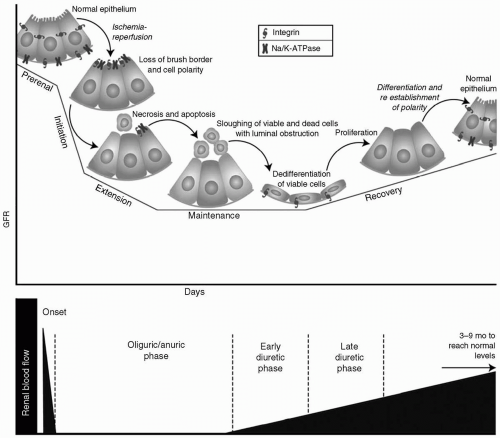
28 Recently, new data emerged that supported the existence of discrete phases of ischemic ATN: Prerenal, initiation, extension, maintenance, and repair (see
Fig. 31.1).
27,
28,
29
Prerenal Phase
Renal hypoperfusion plays a role in the pathogenesis of prerenal azotemia and ATN. In experimental studies, AKI progressed from renal vasoconstriction and intact tubular function (prerenal azotemia) to established AKI with tubular dysfunction if the renal ischemia was prolonged.
30 Moreover, early intervention with fluid resuscitation was shown to prevent the progression from prerenal azotemia to established AKI in some instances.
Although a decrease in RBF with diminished oxygen and substrate delivery to the tubule cells is an important ischemic factor, the relative increase in oxygen demand by the tubules also plays a role in renal ischemia.
30 Although the kidney receives 20% to 25% of cardiac output, most of that flow is directed to the renal cortex, with only a small fraction reaching the vasa recta of the renal medulla.
23 This area of low blood flow influences the concentration of urine but, ironically, places the medulla at risk of further ischemic damage. Unlike the renal cortex that has a partial pressure of O
2 of approximately 50 mmHg, the
outer medulla has a partial pressure of oxygen in the range of 10 to 20 mmHg.
31 Perturbations in medullary blood flow and oxygen delivery can, therefore, lead to anoxic injury if they exceed the critical thresholds when they are not reversed by timely therapy. Cell injury and ATN can result.
23 The precise threshold for this injury and its clinical measurement are unclear. Persistent inadequate blood flow to the kidneys and progression to AKI following successful resuscitation and restored global hemodynamic parameters in severe sepsis and septic shock clearly indicate that the current means for measurements of end-organ perfusion, including that of the kidneys, are inadequate.
32
Initiation Phase
After the initial insults, parenchymal injury develops, and kidney function may deteriorate. Experimental and human studies indicate that tubular epithelial cells can suffer one of three distinct fates after ischemic AKI: Cell death (apoptosis), sublethal cell injury, or escape from injury.
26Apoptosis occurring in both distal and proximal tubular cells is emerging as the major mechanism of early and late cell death in ischemic and nephrotoxic forms of AKI. Necrosis of the tubular cells is restricted to the highly susceptible, outer medullary regions following severe renal injury.
33 However, most cells remain viable, either entirely escaping injury or undergoing sublethal injury and subsequent recovery. Severe reduction in RBF induces characteristic, rapidly occurring, and duration-dependent effects in numerous renal cell types, although these effects are seen most prominently in proximal tubular cells.
34,
35,
36In sublethal tubule cellular injury, alterations in the apical cytoskeleton result in loss of the brush border and cell polarity, and produce disruption of tight and adherens junctions.
26,
34 Loss of basolateral Na
+/K
+-ATPase impairs proximal tubular sodium reabsorption, with a consequent increase in the fractional excretion of sodium.
37 Redistribution of integrins to the apical membrane results in detachment of viable cells from the basement membrane and promotes tubular obstruction.
26 Loss of surface membrane, tubular obstruction resulting from sloughing of viable and dead tubular cells into the lumen, and cast formation decrease the glomerular filtration rate (GFR).
30,
38 Obstruction of the tubular lumen eliminates glomerular filtration within that nephron by increasing intratubular pressures to levels inconsistent with filtration.
34Unfortunately, human studies using forced diuresis with furosemide or mannitol fail to demonstrate an impact on the survival and renal recovery in patients with AKI, indicating that obstruction alone accounts only for minor dysfunction.
39,
40 Tubular “backleak” of glomerular filtrate into the circulation results from denudation of the tubular basement membrane, intratubular obstruction, loss of cell polarity, and disruption of tight and adherens junctions, further contributing to the decline of GFR in AKI.
30Several important mechanisms underlie changes in the tubular cell structure. A profound reduction in intracellular adenosine triphosphate (ATP) content occurs early after ischemic renal injury, leading to several critical metabolic consequences. A rapid degradation of ATP results in the accumulation of adenine nucleotides and hypoxanthine, contributing to the generation of reactive oxygen molecules that cause renal tubule cell injury by oxidation of proteins, peroxidation of lipids, damage to DNA, and induction of apoptosis.
26 A rise in free intracellular calcium causes protease and phospholipase activation, with resultant cytoskeletal degradation.
30 Ischemic activation of nitric oxide (NO) synthase and NO generation in tubule cells further promotes cellular damage through oxidant injury, as well as protein nitrosylation.
41 Even with resolution of the initial ischemic insult, a steady decline in RBF continues, caused by hemodynamic and vascular abnormalities that are incompletely understood.
28,
42 Tubuloglomerular feedback may be partly responsible for the renal vasoconstriction observed in AKI, although its role is not completely clear.
26,
42A complex cascade of inflammatory mechanisms is initiated in this phase and will become fully evident if the injury is not alleviated. Intrarenal protective mechanisms, such as the induction of heat shock protein (HSP) in tubular cells, are activated early and in parallel with mechanisms of renal injury. If they prevail, the injury will be diminished at this stage, with complete recovery to follow.
43,
44
Extension Phase
Extension of microvascular endothelial injury resulting in the perpetuation of hypoxia following the initial ischemic event and exaggeration of inflammatory cascade are the hallmarks of this stage. Regional alterations in RBF persist after the initial ischemic event and are of greater magnitude in the outer medulla than in the outer cortex or inner medulla. Medullary blood flow remains severely reduced to levels ranging from 10% to 50% of normal in both human
28 and animal models of ischemic AKI.
45 The mechanisms underlying this persistent alteration of renal perfusion following ischemic injury are complex and not completely understood. An imbalance between the mediators of renal vasoconstriction and renal vasodilatation, as well as congestion of the renal microcirculation secondary to endothelial injury and dysfunction, are among the major proposed mechanisms.
27,
29Endothelial cells undergo an array of complex changes that alters their function and interaction with multiple other cell types. Endothelial cell swelling, altered cell-to-cell attachment, and reduced endothelial cell basement membrane attachment increase permeability and interstitial edema.
29,
34 Thrombin activation and generation lead to an activated coagulation cascade, and the amplification of inflammatory cascade includes white blood cell activation and cytokine generation.
27 Altered vascular reactivity, produced by reactive, oxygen species-mediated decreases in NO-mediated vasodilatation and endothelin-mediated vasoconstriction, potentiate the intense vasoconstriction and contribute to abnormal blood flow.
29 These changes are most prominent in the peritubular capillaries of the corticomedullary junction and outer medulla, and further exacerbate medullary congestion, hypoperfusion, and injury during the extension phase.
27More widespread tubular cell death, desquamation, and luminal obstruction continue in the medulla. Injured endothelial and epithelial cells amplify the inflammatory cascade and produce multiple inflammatory cytokines and chemokines. Endothelial activation enhances endothelial-leukocyte interactions and leads to leukocyte infiltration, as well as further inflammatory and cytotoxic injury.
27 In a recent study, CD4
+ T cells, working through both interferon-γ (IFN-γ) and costimulatory molecules, were implicated as an important modulator of AKI.
46 Complement activation may also contribute to neutrophil recruitment and tissue damage.
47 This activated inflammatory cascade exacerbates hypoperfusion in the renal microcirculation and promotes further release of cytotoxic inflammatory cytokines and reactive oxygen species.
Maintenance Phase
During this phase, GFR is maintained at its nadir while parenchymal injury is established. RBF begins
to normalize, although it may remain slightly below normal 12 to 20 weeks after the onset of ATN in spite of clinical renal recovery.
28 Tubular cellular injury with ongoing apoptosis coexists with regeneration during the maintenance phase, the duration and severity of which may be determined by the balance between cell survival and death.
44 Repair of both epithelial and endothelial cells is critical to overall recovery, and measures to accelerate the endogenous regeneration processes may be effective during this phase.
Although resident stem cells with tubulogenic capacity have been described in the kidneys after ischemic AKI,
48 proliferation of the native tubule epithelial cells seem to represent the primary healing source for regeneration of the tubule epithelium.
49,
50 The surviving renal tubule cells recapitulate phases and processes that are very similar to those during normal kidney development.
44,
51 Viable cells dedifferentiate, proliferate, and migrate across the basement membrane to reestablish epithelial continuity. This sequence is followed by the process of redifferentiation and reestablishment of normal epithelial polarity and transport functions.
44,
51 Although the beneficial effect of bone marrow-derived, mesenchymal stem cells in ATN recovery has been demonstrated, most of the evidence suggests that this protective effect is mediated through powerful anti-inflammatory and antiapoptotic mechanisms, with decreased renal tubular injury, rather than by differentiation.
52,
53
Recovery Phase
Improvement in GFR and reestablishment of tubular integrity, with full differentiation and polarization of regenerated epithelial cells, are observed in this phase. The mechanisms whereby most of the tubular cells escape cell death and recover completely after ischemic AKI are still under investigation. Induction of HSP is a highly conserved, innate cellular response and is found to be activated after ischemic AKI, likely promoting cell survival by inhibiting apoptosis.
43 The importance of inducing protein involved in cell cycle control has recently emerged.
54 Engagement of the cell cycle is an important determinant of whether cells survive the injury itself. Coordinated cell cycle control, initially manifested as cell cycle inhibition, is necessary for optimum recovery from ATN. Hence, the importance of cell cycle inhibitors, p21 and 14-3-3r, induced by cellular stress from the initial ischemic or nephrotoxic insult, and their combined activities to check the cell cycle at G1 and G2, can be appreciated to coordinate the cell cycle and permit cell survival.
54In conclusion, after the initial “functional recovery” from an episode of clinical AKI, a significant proportion of patients exhibit persistent or progressive deterioration in renal function. Animal studies, and some early morphologic studies in human ATN, indicate that postischemic kidneys demonstrate various sequelae of acute injury, including reduction in renal microvasculature, interstitial fibrosis, tubular hypercellularity and atrophy, and persistent inflammation.
28,
55,
56,
57,
58 Future research efforts will be directed toward a better understanding of the mechanisms described and the development of a means for intervention early in the course of AKI. Perhaps more importantly will be the development of strategies to identify AKI susceptibility among individuals exposed to known ischemic and nephrotoxic injuries and to prevent injury altogether.