I. DEFINITION
A. The RIFLE criteria (Risk, Injury, Failure, Loss, and ESRD) were developed in 2004 to standardize the definition of acute kidney injury (AKI), formerly called acute renal failure (ARF). Prior to this, no consensus was available on the diagnosis or degree of severity.
1. Several modifications were introduced by the Acute Kidney Injury Network (AKIN) soon after, though the main addition with AKIN was a more inclusive Stage 1 (≥0.3 mg/dL increase in Cr).
2. In 2012, the Kidney Disease Improving Global Outcomes (KDIGO) organization published clinical practice guidelines to create a unified definition with the goal to improve outcome staging and future clinical research (since RIFLE and AKIN did not completely coincide).
3. Although each of these (see Table 23.1) were created to help standardize clinical outcomes research, they are helpful when assessing severity of injury and level of management (Fig. 23.1).
4. Serum creatinine criteria: well validated, but discrepancies exist between various definitions (e.g., misclassification of AKI using AKIN with postsurgical ICU patients after cardiopulmonary bypass with significant positive fluid balance resulting in hemodilution).
5. Urine volume criteria are the same for all three (RIFLE/AKIN/KDIGO) but oftentimes it is less accurate (e.g., morbid obesity).
6. Validation studies are currently underway to establish the utility of these guidelines—particularly with regard to diagnosis and outcome.
II. EPIDEMIOLOGY
A. The historical lack of a standardized definition results in variable epidemiological data for AKI. Information bias and residual confounding (e.g., variability in baseline measures, the use of diagnostic code results), as well as ascertainment bias (AKI vs. CKD, which have different pathophysiologic processes and outcome data), results in differences in reported incidence and outcome measurements.
B. Even with the above definitions, studies demonstrate significant differences in the incidence of AKI in the ICU when comparing the same populations.
C. Nonetheless, AKI is common in both hospitalized (5%–20%) and critically ill (30%–40%) patient populations.
D. Higher level of severity in AKI classification (by any of the above definitions) is associated with adverse outcomes, including increased length of stay, progressive kidney disease, and mortality.
E. AKI is an independent risk factor for cardiovascular complications and mortality; AKI requiring renal-replacement therapy reveals an in-hospital mortality of 50% to 75%. Studies have revealed that up to 28% of surviving AKI patients died after discharge from the hospital (i.e., in-hospital mortality likely underestimates the significance of disease).
F. AKI patients often regain renal function with supportive therapy; however, studies have demonstrated more severe AKI, longer AKI duration, and numerous episodes of AKI are associated with progression to CKD and increasing morality. Future insults are much less well tolerated in these patient populations (e.g., additive effect of renal injuries over time).
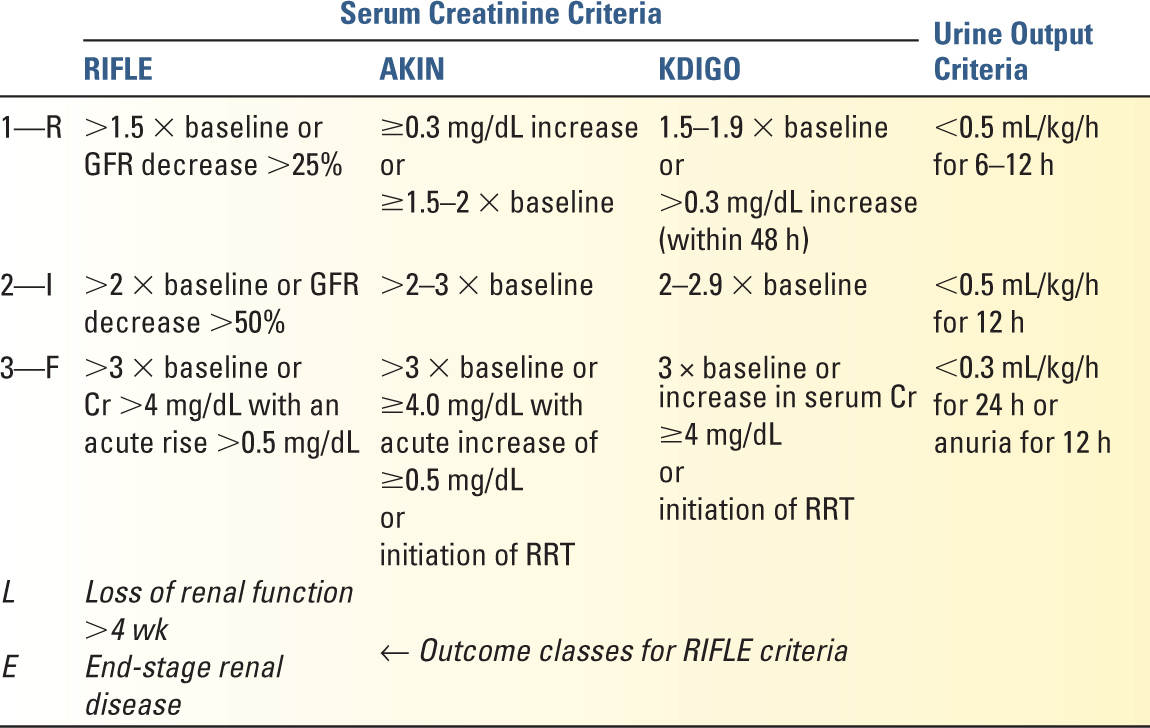

FIGURE 23.1 Stage-based management of AKI. Shading of boxes indicates priority of action—solid shading indicates actions that are equally appropriate at all stages whereas graded shading indicates increasing priority as intensity increases. AKI, acute kidney injury; ICU, intensive-care unit. (From Kidney Disease: Improving Global Outcomes [KDIGO] Acute Kidney Injury Work Group. KDIGO clinical practice guideline for acute kidney injury. Kidney Int Suppl 2012;1(2):1–138.)
III. ETIOLOGY AND PATHOPHYSIOLOGY
AKI is often caused by more than one etiology, but the pathophysiologic processes can be separated into three separate classifications: prerenal, intrinsic (or “renal”), and postrenal injuries. This is helpful both to determine the underlying etiology and for management (Table 23.2).
A. Prerenal (Kidney Hypoperfusion)
1. Decreased intravascular volume
a. Systemic vasodilation (e.g., sepsis). Sepsis is the most common cause of AKI in the ICU (approximately 50% can be attributed) and is associated with a very high mortality.
b. Hypovolemia (e.g., hemorrhage, GI losses, burns)
c. Hypotension, decreased cardiac output (CHF, arrhythmias)
d. Small vessel (renal) vasoconstriction
1. Nonsteroidal anti-inflammatory drugs (NSAIDs): Prostaglandins have important vasodilatory effects on the afferent arteriolar vessels. Cyclooxygenase inhibitors inhibit the production of prostaglandins, resulting in decreased renal blood flow.
2. ACEi/ARBs: Angiotensin-II is a potent efferent arteriolar vasoconstrictor, and the inhibition of its production (ACE inhibitors) or the blockage of angiotensin receptors (ARBs) results in decreased glomerular perfusion pressure. This is of particular concern when the patient has other risk factors (chronic kidney disease, renal artery stenosis, older age) or exposures (diuretics, hypotension, NSAIDs, nephrotoxins).
3. Contrast: IV contrast is both cytotoxic to renal tubular cells and causes intrarenal vasoconstriction.
4. Hypercalcemia: directly causes vasoconstriction
5. Calcineurin inhibitors (CNIs): Both cyclosporine and tacrolimus, which lead to afferent and efferent arteriolar vasoconstriction, are used as immunosuppressants for patients after renal transplant.
6. Hepatorenal syndrome (HRS): Splanchnic and systemic vasodilation causes increased neuroendocrine (angiotensin, vasopressin) tone, resulting in renal vasoconstriction. Prognosis is very poor.
7. Intra-abdominal hypertension (IAH; IAP ≥12 mmHg) and abdominal compartment syndrome (ACS; IAP ≥20 mmHg with new organ failure): The pathophysiology is likely a combination of (initially) intrarenal venous congestion/hypertension (normally a low-pressure system), followed by decreased cardiac output and elevated catecholamines, neurohormones, and cytokines (inflammation). Similar to cerebral perfusion pressure (CPP), abdominal perfusion pressure (APP) is a helpful marker of renal perfusion, where APP = MAP – IAP (typical target APP ≥60 mmHg).
8. Cardiorenal syndrome: In addition to decreased cardiac output (acute CHF) resulting in decreased intravascular volume, other pathophysiological processes are likely involved. Mechanisms likely include a combination of venous congestion (elevated CVP) and visceral edema resulting in decreased abdominal perfusion pressure (APP) and elevated neuroendocrine (angiotensin, vasopressin) hormones, resulting in decreased renal perfusion.
| Etiologies of Acute Kidney Injury in the Intensive Care Unit | |
Prerenal | Intrinsic Renal | Postrenal (Obstructive) |
Intravascular volume depletion • GI fluid loss (e.g., vomiting, diarrhea, EC fistula) • Renal fluid loss (e.g., diuretics) • Burns • Blood loss • Redistribution of fluid (e.g., “third-spacing,” pancreatitis, cirrhosis) | Acute tubular necrosis • Ischemic • Toxin-induced • Drugs • IV contrast • Rhabdomyolysis • Massive hemolysis • Tumor lysis syndrome | Upper urinary tract obstruction • Nephrolithiasis • Hematoma • Aortic aneurysm • Neoplasm |
Decreased renal perfusion pressure • Shock (e.g., sepsis) • Vasodilatory drugs • Preglomerular (afferent) arteriolar vasoconstriction • Postglomerular (efferent) arteriolar vasodilation | Acute interstitial nephritis • Drug-induced • Infection-related • Systemic diseases (e.g., SLE) • Malignancy | Lower urinary tract obstruction • Urethral stricture • Hematoma • Benign prostatic hypertrophy • Neurogenic bladder • Malpositioned urethral catheter • Neoplasm |
Decreased cardiac output • Congestive heart failure • Myocardial ischemia | Acute glomerulonephritis • Postinfectious • Systemic vasculitis • TTP/HUS • Rapidly progressive GN | |
| Vascular • Atheroembolic disease • Renal artery or vein thrombosis • Renal artery dissection • Malignant hypertension |
|
| Hepatorenal syndrome Increased intra-abdominal pressure |
|
GI, gastrointestinal; EC, enterocutaneous; IV, intravenous; SLE, systemic lupus erythematosus; TTP, thrombotic thrombocytopenic purpura; HUS, hemolytic uremic syndrome; GN, glomerulonephritis.
Adapted with permission from Barozzi L, Valentino M et al. Renal ultrasonography in critically ill patients. Crit Care Med 2007;35(5 suppl):S198–S205 and Acute renal failure. In: Glassock RJ, ed. Nephrology self-assessment program (NephSAP), Vol. 2. Philadelphia: Lippincott Williams & Wilkins, 2003:42–43.
e. Large-vessel etiologies
1. Renal artery stenosis (RAS): This usually is in combination with other etiologies (e.g., ACE inhibitors, hypotension).
2. Aortic or renal artery dissection (or compression)
3. Thrombosis or embolism of the renal vessels
B. Intrinsic Renal Injury
1. Acute tubular necrosis (ATN)
a. Ischemia (prerenal azotemia naturally progresses into ATN): This is the most significant cause of ATN in the intensive care unit.
b. Pigments (hemoglobin, myoglobin): Of note, rhabdomyolysis should be considered with CPK (creatinine phosphokinase) levels >5,000 units/L. This frequently results in hyperkalemia, hyperphosphatemia, hyperuricemia, hypocalcemia, and acidosis. Injury is secondary to direct tubular toxicity from cast formation, in addition to intrarenal vasoconstriction and hypovolemia (muscle inflammation and third spacing). It is important to remember that CPK levels are a marker for muscle injury, and direct injury is caused by myoglobin.
c. Warfarin-related nephropathy (WRN) is a recently described entity where an elevated INR (>3.0) places patients at risk for hematuria and erythrocyte occlusion in Bowman’s space and renal tubules (erythrocyte casts present on biopsy). Further investigation is underway.
d. Drugs
1. Aminoglycosides, particularly at high doses, are toxic to proximal tubular cells. Once-daily dosing and vigilant dosing of medications can prevent some but not all cases.
2. Amphotericin B can cause AKI due to nephrotoxicity and vasoconstriction. It is also associated with the development of a distal RTA. Liposomal and colloid dispersion may decrease the incidence of severe AKI.
3. Vancomycin has been shown to be associated with ATN; however, the incidence has decreased since the preparation has changed. Higher doses/levels may lead to increased incidence.
4. Hydroxylethyl starches (HES) are associated with significant AKI, and increased mortality, when used in the critically ill patients with sepsis as a volume expander. Osmotic nephrosis is seen in the proximal tubules.
5. Contrast-induced AKI (CIAKI) or contrast-induced nephropathy (CIN): Contrast is both directly cytotoxic and decreases renal blood flow by vasoconstriction. This usually is in combination with hypovolemia/hypotension.
e. Proteins (immunoglobulin light chains—multiple myeloma): Notably, intravenous immunoglobulin (IVIG) therapy has been associated with proximal tubular osmotic nephrosis and possibly arterial vasoconstriction.
f. Crystals (uric acid, acyclovir, methotrexate)
2. Acute interstitial nephritis (AIN)
a. Allergic (drug-induced): The most common drugs include β-lactam antibiotics and sulfonamides. Others include NSAIDs, PPIs, fluoroquinolones, vancomycin, and phenytoin.
b. Infection: Bacterial and viral infections can lead to AIN by direct (pyelonephritis) and indirect (legionella) renal involvement.
c. Infiltrative/autoimmune: Sarcoidosis, lupus, lymphoma/leukemia, and other systemic diseases can lead to interstitial inflammation.
3. Glomerulonephritis
a. Intraglomerular inflammation with various timelines of progression and acuity of disease: Progression can be acute or rapidly progressive and severe requiring early diagnosis. Pulmonary hemorrhage is a rare but feared complication (can be seen with ANCA or anti-GBM disease). Typically classified on the basis of pathology, that is, pauci-immune (ANCA vasculitis), anti-GBM, and immune complex (postinfectious, IgA, lupus nephritis, membranoproliferative, cryoglobulinemia, etc.).
4. Small vessel disease
a. Thrombosis: various pathophysiological processes including hemolytic uremic syndrome (HUS), thrombotic thrombocytopenic purpura (TTP), preeclampsia, antiphospholipid antibody syndrome (APLAS), polyarthritis nodosa (PAN), scleroderma, and disseminated intravascular coagulopathy (DIC)
b. Cholesterol embolism
C. Postrenal (Obstruction and Hydronephrosis)
1. Bilateral ureteral obstruction
a. Malignancy (including lymphadenopathy), aneurysmal compression, nephrolithiasis, or retroperitoneal fibrosis or hematoma
2. Bladder/urethral pathology
a. Prostate enlargement (from benign prostatic hyperplasia or malignancy), Foley catheter or urethral obstruction, anticholinergic medications, and neurogenic bladder can all contribute to urinary obstruction and postrenal kidney dysfunction.
IV. EVALUATION
A. History should focus on baseline kidney function, risk factors (see Sections V.A and V.B), including medications, comorbidities, and recent procedures or events.
B. Physical examination to include vital signs and volume status (intravascular volume), bedside focused ultrasonography (e.g., inferior vena cava diameter), and signs/symptoms of vascular disease
C. Renal Function Evaluation
1. BUN (nonspecific; increased with gastrointestinal bleeding, steroids, high-protein tube feeds, hypermetabolism—especially with proteins)
2. Serum creatinine (Cr; affected by muscle mass, amputation, drugs, and volume status)
3. 24-hour creatinine clearance (can also check if more rapid results are desired)
4. Urine output (not sensitive)
5. Biomarkers of AKI (NGAL, KIM-1, cystatin C, etc.) might provide future early AKI diagnosis; however, prospective trials are necessary before these are clinically applicable.
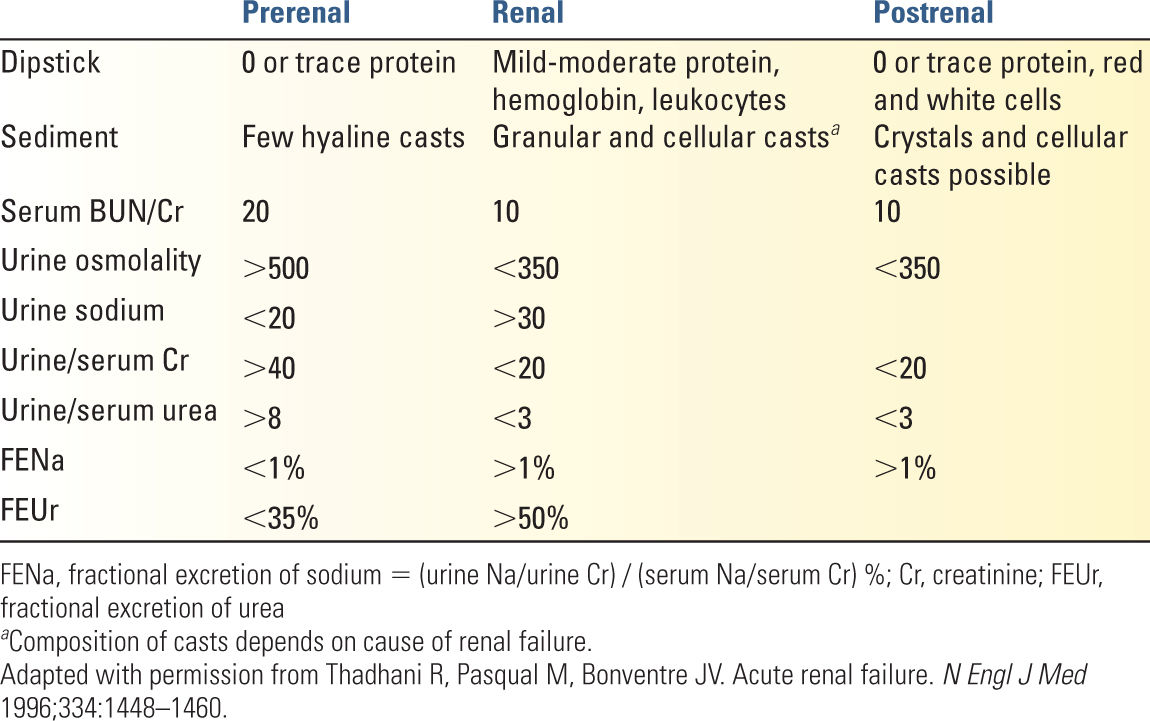
6. Urine evaluation (Table 23.3)
a. Urinalysis and sediment
1. Prerenal: bland urinalysis and (transparent) hyaline casts
2. Intrinsic etiologies demonstrate:
a. Granular (muddy brown) casts (ATN secondary to ischemia or nephrotoxic injuries) can also be seen in the setting of glomerular disease.
b. Pigmented casts (ATN secondary to hemoglobin or myoglobin or with hyperbilirubinemia)
c. RBC casts (glomerulonephritis)
d. WBC casts (pyelonephritis, AIN, or glomerulonephritis)
e. Urine eosinophils (AIN, cholesterol emboli, though not very sensitive or specific)
3. Postrenal: bland urinalysis. RBCs with nephrolithiasis
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


