
ACUTE KIDNEY INJURY
The functions of the kidney can be divided into secretion of hormones and maintaining the extracellular homeostasis of pH, water, and electrolytes. The kidneys weigh approximately 300 g each but receive blood flow disproportionately, approximately 20%–25% of cardiac output. The kidneys consume more oxygen per gram of mass than any other visceral organ. The kidneys secrete renin, erythropoietin, the active form of vitamin D, and certain prostaglandins. Most importantly, the kidneys regulate fluid balance, acid–base status, and solute homeostasis.
After passing through the interlobar, arcuate, and intralobar arteries, more than 85% of renal blood flow perfuses the outer cortical glomeruli, leaving the remainder to supply the juxtamedullary glomeruli.1
The kidney’s ability to perform many of its functions depends on three fundamental functions: filtration, reabsorption, and secretion at the level of the nephron. The nephron, the smallest functional unit of the kidney, includes a renal corpuscle, which is composed of a glomerular capillary tuft enclosed in Bowman’s capsule. The glomerulus is distinct from other capillary beds in that it is situated between two muscular arterioles instead of between an arteriole and a venule. The afferent arteriole feeds into the glomerular tuft; its tone is modulated principally by a myogenic reflex, which adjusts arteriolar tone in response to perfusion pressure, and by certain prostaglandins. The efferent arteriole drains the tuft; its tone is largely determined by activity of the renin–angiotensin system. By dynamic adjustment of the tone of these two arterioles, renal blood flow and glomerular filtration pressure are autoregulated over a wide range of systemic blood pressures. Cells, proteins, and other large molecules entering the glomerulus are excluded from filtration subsequently by a fenestrated endothelium and a size and charge-specific glomerular basement membrane. However, smaller molecules, electrolytes, and plasma fluid can pass into the Bowman space by ultrafiltration, which is driven by the capillary hydrostatic forces described above. This remaining effluent, or ultrafiltrate, then passes through the proximal tubule, the loop of Henle, the distal convoluted tubule, and finally the collecting ducts.2 During this process, fluid is modified for water and solute regulation; the resultant fluid is urine.
The proximal tubule can be divided into an initial convoluted portion and a more distal descending portion, and functions primarily to reabsorb filtered plasma components. Fluid in the filtrate entering the proximal convoluted tubule is reabsorbed into the peritubular capillaries with approximately two-thirds of the total filtered salt and water and all the filtered organic solutes, which include primarily glucose and amino acids.3 A substantial portion of bicarbonate reclamation also occurs here, largely due to the function of carbonic anhydrase.
The loop of Henle is a U-shaped conduit that extends from the proximal tubule and consists of descending and ascending limbs. It begins in the cortex, receiving filtrate from the straight portion of the proximal tubule, and extends into the medulla as the descending limb, before it returns to the cortex as the ascending limb to empty into the distal convoluted tubule. Its primary roles are to reabsorb 25%–35% of the filtered NaCl load via the Na+K+2Cl– loop diuretic–sensitive transporter, and to create gradients that allow for elaboration of either dilute or concentrated urine, depending on the immediate physiologic need.4 The descending limb is permeable to water but impermeable to salt, whereas the ascending limb is impermeable to water. In the descending limb, the hypertonic interstitium of the renal medulla (established via active transport of sodium into the interstitium in the ascending limb) causes water to flow freely out of the descending limb by osmosis until the tonicity of the filtrate and interstitium equilibrate. The ascending limb pumps sodium continuously out of the filtrate, perpetuating a hypertonic interstitium and hypotonic tubular fluid. This process is called countercurrent multiplication, and results in an increasingly concentrated tubular fluid and interstitium at the bottom of the loop of Henle and a progressively more dilute filtrate by the start of the distal convoluted tubule.4 The hypertonic interstitium eventually provides the osmotic gradients to allow water reabsorption later in the nephron, whereas the newly hypotonic filtrate (~70 mOsm/L) can be diluted further in the distal convoluted tubule if free water excretion is desired.
The distal convoluted tubule reabsorbs more salt via the NaCl thiazide-sensitive channel and, in the absence of vasopressin (antidiuretic hormone, ADH), can dilute the urine to approximately 40 mOsm/L in normal kidneys. The distal convoluted tubule is also involved in calcium reabsorption and phosphate excretion under the influence of parathyroid hormone. The filtrate then passes to the collecting tubules, where final fine-tuning of acid–base, sodium, and potassium balance occur, largely under the influence of aldosterone. The cortical and medullary collecting tubules are also the main site of ADH-directed water reabsorption. Here, the highly concentrated interstitium derived from active salt transport in the loop of Henle is used to drive passive water reabsorption. Thus, from normal kidneys, urine as dilute as 40 mOsm/L and as concentrated as 1,200 mOsm/L can be elaborated based on bodily needs.4
OVERVIEW OF ACUTE KIDNEY INJURY
Acute kidney injury (AKI), or acute renal failure as the entity was called previously, remains a common and lethal complication of critical illness. The literature is replete with definitions of AKI, but the occurrence of AKI in a patient is most often defined as an elevation of serum creatinine concentration (s[Cr]) of 0.5 mg/dL from baseline, or as an acute need for renal replacement therapy (RRT), for example, hemodialysis.5 The lack of consensus on the definition of AKI has affected the ability of clinicians to compare published protocols regarding diagnosis and treatment of this prevalent disorder. As such, a workgroup called the Acute Dialysis Quality Initiative developed a consensus definition of AKI (Table 52.1) incorporating risk, injury, failure, loss, and end-stage renal failure criteria (“RIFlE”) for the individual patient.6 These criteria account for either elevation of s[Cr] or decrements of glomerular filtration rate (GFR) or urine output in the determination of the different stages of AKI. These criteria also yield prognostic information regarding the status of the critically ill patient. However, although these criteria are well accepted in the research community with virtually all AKI publications using the RIFlE criteria by 2006,7 adoption in routine clinical use remains uncommon.
TABLE 52.1
RIFLE CRITERIA
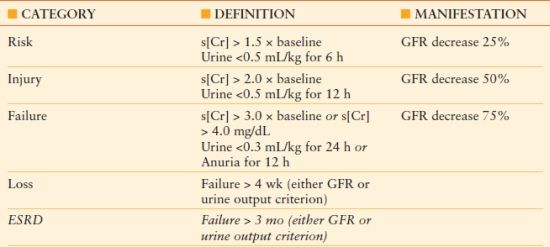
s[Cr], serum creatinine concentration; ESRD, end-stage renal disease; GFR, Glomerular filtration rate; RIFLE, risk, injury, failure, loss, and end-stage renal disease criteria
The occurrence of AKI may influence profoundly the outcome of critically ill patients.8 In a study of postoperative cardiac patients, patients with even the slightest augmentations of s[Cr] that qualified them for RIFlE R criteria of AKI had a 2.2-fold greater mortality, a 1.6-fold increase in ICU length of stay, and 1.6-fold increase in total postoperative costs compared to controls.9 Similarly, a meta-analysis encompassing 78,855 patients found that even just a 10%–24% increase in s[Cr] was associated with an 80% increase in mortality compared with matched patients and that a s[Cr] increase >50% was associated with an eightfold increase in mortality.10 Therefore, even small changes in s[Cr] should be of concern, and should prompt careful evaluation of potential causes and treatments.
Clinical Syndromes of AKI
The etiology of AKI is multifactorial in most patients and may represent only a portion of a patient’s organ dysfunction. Whereas several etiologies may lead to AKI, each individual contributor to AKI is generally classified as being prerenal, “intrinsic” renal, or postrenal (Table 52.2).
TABLE 52.2
ETIOLOGIES OF AKI

Prerenal Azotemia. “Prerenal” AKI is believed not to be due to an actual change in the kidneys’ ability to clear Cr and other solutes, but rather to a perfusion-limited decrease in solute presentation to the kidneys. Most commonly, this is due to intravascular volume depletion, such as might occur during hemorrhage, desiccation, or third spacing of intravascular fluid. However, it should be noted that “prerenal” does not necessarily mean a given patient is whole body, or even volume depleted intravascularly. Congestive heart failure, for instance, is often associated with “prerenal” increments in s[Cr], and is due to inability of the heart to generate cardiac output to perfuse the kidneys adequately. As such, prerenal azotemia is best characterized by ineffective arterial blood flow to the kidneys for any reason. AKI from prerenal causes is usually reversible, and will generally respond to correction of the underlying abnormality (i.e., fluid resuscitation for intravascular volume depletion).
Intrinsic Acute Kidney Injury. Structural injury of the kidney is the hallmark of intrinsic AKI (Table 52.2). The most common form is acute tubular necrosis (ATN), whether ischemic due to progression of prerenal azotemia or interruption of renal blood flow (e.g., trauma, thromboembolism) or cytotoxic (e.g., due to toxicity from medications or ingestion syndromes).11 ATN may occur even absent an overt, prolonged decrease in renal blood flow or a massive exposure to nephrotoxins, usually due to a confluence of events in the acute setting. For example, doses of gentamicin insufficient to cause AKI in a normal kidney may result in substantial nephrotoxicity in the setting of hypoperfusion or concurrent exposure to other nephrotoxins.12 However, ATN occurs most commonly when the nephrons’ capacity to autoregulate blood flow is overwhelmed. As noted above, this autoregulation is dependent on modulation of the relative tones of the glomerular afferent and efferent arterioles. Moreover, oxygen tension in the renal medulla tends to be low, due to both the anatomic characteristics and energy demands of the loop of Henle. The medullary microcirculation is dependent on endothelial elaboration of vasodilators such as nitric oxide and prostaglandins, which may be impaired in acute illness, existing microvascular disease, or by medications such as radiocontrast medium or nonsteroidal anti-inflammatory drugs (NSAIDs).13 Thus, the stressed renal microvasculature loses its autoregulatory capacity and becomes more sensitive to vasoconstrictor drugs and periods of systemic hypotension, which thus may provoke additional damage that can delay recovery from ATN.14 Although the cellular injury is observed in proximal tubules predominantly (by contrast to apoptotic cell death, which occurs primarily in the distal nephron), necrosis of the distal nephron may also occur. The distal nephron may also become obstructed by desquamated cells and cellular debris due to sloughing of more proximal tubular cells. Endogenous growth factors regulate tissue repair and recovery; administration of growth factors exogenously has been shown experimentally to ameliorate and hasten recovery from AKI, although there have been no studies suggesting benefit in clinical trials. Depletion of neutrophils and blockade of neutrophil adhesion reduce AKI following experimental ischemia, indicating that inflammation is at least partly responsible for some features of ATN, especially in postischemic injury after transplantation.14
Hypoperfusion-related decreases in glomerular blood flow and intrarenal vasoconstriction appear to be the dominant mechanisms for reduced GFR in ATN. The mechanisms of this vasoconstriction are not entirely clear, but tubuloglomerular (TG) feedback mechanisms appear to play a role. In the normal setting, TG feedback can modulate glomerular blood flow by causing arteriolar vasoconstriction when large amounts of chloride are presented to the macula densa, a group of cells at the end of the loop of Henle that physically abut its associated glomerulus.4 This response is favorable from an evolutionary perspective; when there is tubular dysfunction, defects in salt and chloride reabsorption lead to activation of TG feedback and a resultant decrease in blood flow to the affected glomerulus. Salt and water losses are thereby mitigated, at the cost of some degree of GFR. Tubular obstruction from sloughed cells and interstitial edema also result in loss of the hydrostatic gradients favoring glomerular filtration.
Whereas ATN comprises the large majority of “intrinsic” causes of AKI, two other less common causes are due to host immune responses: postinfectious glomerulonephritis (PIGN) and acute interstitial nephritis (AIN). The incidence of PIGN in the acute surgical setting has not been studied but is believed to be rare, although that impression may be due to a paucity of biopsy data in this population. Although the pathophysiology of PIGN has not been elucidated, it is conceptualized as an immune complex–associated glomerulonephritis, in which antigen–antibody complexes containing bacterial (usually streptococcal) antigen deposit in the glomerulus and cause immune activation and inflammation.15 This can result in varying degrees of renal dysfunction, from mild loss of GFR to a permanent need for RRT. Common features of PIGN include signs of acute kidney inflammation, including salt retention and edema, hypertension, hematuria, hemolysis, and proteinuria.
Interstitial nephritis is also an important contributor to intrinsic AKI. Predominantly due to allergic responses to medications, AIN was implicated as the cause in 7% of cases of AKI in a recent biopsy review.16 Although any medication can theoretically cause AIN, the most common culprits include penicillins (especially nafcillin or methicillin [the latter now used rarely]), cephalosporins, NSAIDs, sulfa-containing medications, rifampin, fluoroquinolones, and proton pump inhibitors.17 Time of onset of AKI is unpredictable, and can occur from 1 day to months after the second exposure to the agent. As such, awareness of and suspicion of AIN is important, especially in the setting of known potential medications, peripheral or urine eosinophilia, and rash.
Postrenal Acute Kidney Injury. “Postrenal” AKI is due to mechanical obstruction of urine flow, which results not only in failure to excrete urine, but also the eventual loss of the hydrostatic gradient favoring filtration across Bowman’s capsule if the obstruction is not relieved (Table 52.2). Obstruction can occur anywhere along the urinary tract, including the ureters, bladder, and urethra. Bladder outlet obstruction due to prostate hyperplasia is the most common cause of postrenal AKI. Unilateral causes of hydronephrosis, such as a ureteral calculus or extrinsic compression of one ureter, generally do not lead to oliguria, as the contralateral kidney will continue to produce urine. As such, whereas placement of a Foley catheter does essentially rule out bladder outlet obstruction (or overcome it if present), bladder catheterization does not rule out obstructive nephropathy more proximally. In addition, although not truly “obstructive” in nature, postoperative urine leaks can also present as “postrenal” AKI. Urine leaks occur most commonly after surgical manipulation of the bladder or ureters (e.g., cystectomy, kidney transplant surgery), and can present as decreased urine output and persistently elevated or increasing s[Cr] without obvious cause. The diagnosis is usually confirmed by radionuclide imaging that detects renal excretion of tracer into a perinephric or intraperitoneal fluid collection. Alternatively, [Cr] can be measured in a sample of the fluid; if it is higher than s[Cr], the fluid collection is at least partially urine.
DETECTION OF ACUTE KIDNEY INJURY
As noted above, the RIFlE criteria have become the standard for definition of AKI, and have provided consistency to AKI diagnoses (Table 52.1). However, complicating further the evaluation and treatment of AKI is the lack of ideal biomarkers thereof. Currently, the blood urea nitrogen concentration [BUN] and s[Cr] are the tests used most commonly to monitor renal function, and as such, s[Cr] is the main laboratory parameter used in the RIFlE classification. Nevertheless, these measures are clearly imperfect. Urea nitrogen, for example, is an end product of protein metabolism, and does accumulate in the presence of AKI and chronic kidney disease (CKD). However, there are many conditions that can also result in an elevated [BUN] unrelated to renal function, including (1) states of protein catabolism (e.g., corticosteroid therapy, sepsis); (2) excessive protein intake; or (3) increased reabsorption (e.g., dehydration, gastrointestinal bleeding). Conversely, patients who cannot synthesize urea (e.g., liver cirrhosis) or have inadequate protein intake may have misleadingly low BUN concentrations even in the presence of substantial kidney dysfunction.
Although s[Cr] is accepted as the standard test to monitor renal function, it is imperfect. Steady-state s[Cr] is influenced by age, gender, and muscle mass. Severe decrements in renal function can be present even while the s[Cr] remains within the laboratory’s specified “normal range,”18 especially among elderly females. For example, a s[Cr] of 1.2 mg/dL in a 90-year-old female with a body mass of 50 kg may underrepresent GFR, whereas the same s[Cr] in a 25-year-old black male with a body mass of 100 kg may be normal. Formulas such as the Cockcroft-Gault or Modification of Diet in Renal Disease (MDRD) calculations (Table 52.3) were developed to normalize demographic variations in muscle mass. Calculation tools are readily available online, and can be used to estimate renal function with reasonable accuracy. These imperfections are noted not to discourage use of [BUN] and s[Cr] for evaluation of renal function, but rather to highlight potential confounding factors that may have clinical import.
TABLE 52.3
FORMULAS FOR ESTIMATION OF CREATININE CLEARANCE (eCCr) OR GLOMERULAR FILTRATION RATE (eGFR)

The MDRD equation has been validated in patients with CKD, but underestimates the GFR in healthy patients with GFRs over 60 mL/min (From Madaio M. Post-infectious glomerulonephritis. In: Schrier RW, ed. Diseases of the Kidney. Boston, MA: Little Brown; 1997:1579–1594; Prakash J, Sen D, Kumar NS, et al. Acute renal failure due to intrinsic renal diseases: review of 1122 cases. Ren Fail. 2003;25:225–233.) The equations have not been validated in AKI. These MDRD equations are to be used only if the laboratory has NOT calibrated its s[Cr] measurements to isotope dilution mass spectroscopy (IDMS). When IDMS-calibrated [sCr] is used (which is about 6% lower), the above equations should be multiplied by 175/186, or 0.94086. These formulas do not adjust for body mass, therefore, relative to the Cockcroft-Gault formula, they underestimate eGFR for heavy people and overestimate it for underweight people. See text for abbreviations.
Emerging biomarkers may prove useful in the future for early detection of AKI. Potential biomarkers include both serum neutrophil gelatinase-associated lipocalin (NGAL) and cystatin C, and urinary NGAL, interleukin-18 (IL-18), and kidney injury molecule-1 (KIM-1).19 Serum NGAL and cystatin C may be particularly useful in predicting AKI after cardiac surgery.20 These biomarkers may also be important for drug development. With highly sensitive urinary biomarkers of kidney toxicity, drugs that have nonclinical signals of nephrotoxicity can be tested more safely in clinical trials.21
PREVENTION OF ACUTE KIDNEY INJURY
The best management strategy for AKI entails preventing its development. Certain patients have been identified clearly as being at high risk of developing AKI, including postoperative patients or those with sepsis, trauma, or hypovolemia,22 including shock of any etiology. Patient groups also at risk include those receiving any nephrotoxic agents (e.g., iodinated radiocontrast medium, medications), those with CKD due to diabetes mellitus or hypertension, or patients with congestive heart failure, multiple myeloma, chronic infection (e.g., osteomyelitis), or a myeloproliferative disorder. Estimation of GFR using either the Cockcroft-Gault or MDRD GFR equation, or measurement of urine creatinine clearance (CCr) is especially important in such patients, as this may improve risk stratification regarding administration of procedures, medications, or imaging tests. Whereas all hospitalized patients must have their intravascular volume status monitored to some degree, these at-risk patients require particularly close monitoring to ensure that hypovolemia is avoided and that exposure to nephrotoxins is minimized or avoided. Consequently, high-risk patients may need frequent measurements of vital signs, central venous pressure, serum [lactate], urine output, or short-duration (e.g., 8–12 hours) CCr.
Furthermore, medications that are not overtly “nephrotoxic” but that do affect glomerular blood flow, such as angiotensin-converting enzyme inhibitors (ACE-I) and NSAIDs, should be used with caution, especially in populations prone to AKI. For instance, in patients with cardiac or liver disease, or patients who may be volume underresuscitated, prostaglandin-mediated afferent arteriolar dilation or angiotensin II–medicated efferent arteriolar constriction may be crucial in maintaining filtration pressure. Blockade of one or both of these mechanisms with a NSAID or ACE-I can result in an abrupt decrease in GFR.
Several interventions may prevent AKI in certain patient groups. Nondiabetic cardiac patients may have a diminished risk of AKI after cardiac surgery, according to the RIFlE criteria, by maintaining tight perioperative blood glucose control. In one study, nondiabetic patients undergoing cardiac surgery had glucose concentrations controlled by insulin infusion at either a trigger of >140 mg/dL, or in a range of 80–111 mg/dL. The observed overall incidence of postoperative AKI or acute postoperative RRT was lower in the group controlled more tightly, who also demonstrated a lower mortality rate.22 Patients with septic shock appear to be at particularly high risk for AKI. These patients may be severely hypovolemic and may also be subject to nephrotoxicity from cytokine-mediated renal vasoconstriction as part of the proinflammatory response. Notably, delays in antibiotic therapy in this cohort of patients may be particularly predictive of AKI.23
Certain intravenous medications have been suggested to prevent the occurrence of AKI. Historically, low-dose dopamine (1.5–3.0 mcg/kg/minute) has been used most commonly.24 D’Orio et al. demonstrated that dopamine (3 mcg/kg/minute) caused isolated renal vasodilation, resulting in increased diuresis, natriuresis, increased renal blood flow, and enhanced GFR, without activating either alpha or beta-adrenergic receptors.25 The institution of low-dose dopamine in critically ill patients will augment CCr by 25% or more in individual patients,26 but large studies have failed to demonstrate benefit to prevent development of AKI or mitigate it postonset. Marik performed a meta-analysis of 15 trials (970 patients) looking at the absolute change of s[Cr] with the use of low-dose dopamine, and found no difference.27 Bellomo et al., through the Australian and New Zealand Intensive Care Society (ANZICS) Clinical Trials Group, performed the largest multicenter randomized trial of low-dose dopamine (by studying critically ill patients with signs of systemic inflammatory response syndrome and clinical evidence of early AKI). This trial of 328 patients in 23 ICUs found that neither the primary outcome (peak s[Cr]) nor any of the secondary outcomes (hourly urine output, requirement for RRT, duration of mechanical ventilation, duration of ICU or hospital stay, or mortality) were altered among patients treated with low-dose dopamine.28 Currently, low-dose dopamine may be used to promote diuresis in select patients with adequate preload, as many patients do respond with increased urine output. However, low-dopamine does not alter the outcome or avert the development of AKI in critically ill patients.
Fenoldopam, a potent dopamine D1 receptor agonist that increases blood flow to the renal cortex and outer medulla, has also been utilized to prevent AKI, but may induce hypotension. Indications for fenoldopam in critical care are not well defined, but it may be beneficial when hypertension must be treated while preserving renal perfusion. In a study using perioperative fenoldopam with N-acetylcysteine, investigators found the combination abrogated an early postoperative decrease of renal function in cardiac surgical patients with CKD, but did not affect other outcomes.29 A meta-analysis of fenoldopam to prevent AKI in postoperative cardiac patients showed that fenoldopam reduced the need for RRT and perioperative mortality.30 Another meta-analysis of randomized, controlled trials (RCTs) of fenoldopam to prevent or treat AKI in postoperative or intensive care patients demonstrated that mortality and the need for RRT were both decreased by fenoldopam.31 However, the second meta-analysis included only small single-center studies, and doses were variable. Data are insufficient presently to support widespread use of fenoldopam for prevention or management of AKI.
SPECIAL POPULATIONS
Rhabdomyolysis
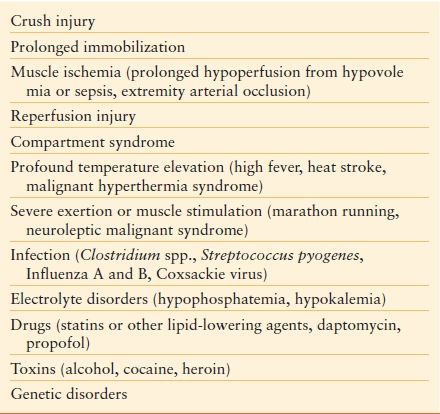
Certain patient populations are at extremely high risk of developing AKI, and warrant special consideration. Rhabdomyolysis is due most commonly to crush injury, but it can also occur with high fever or other clinical syndromes32 (Table 52.4).
TABLE 52.4
Major Etiologies of Rhabdomyolysis
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


