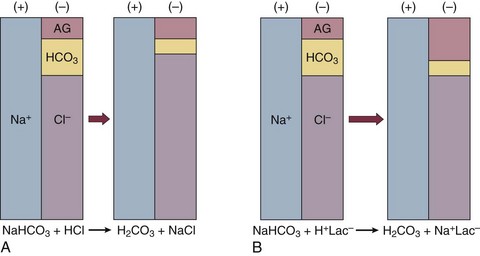
57 Normal biochemical and physiologic function requires that the extracellular pH be maintained within a very narrow range. Although the “normal” range of pH in clinical laboratories is 7.35 to 7.45 pH units, the actual pH in vivo varies considerably less.1 This tight control is maintained by a complex homeostatic mechanism involving buffers and the elimination of volatile acid by respiration. The principal extracellular buffer system is the carbonic acid/bicarbonate pair. The equilibrium relationships of the components of this system are illustrated as follows:1 From these relationships, the Henderson-Hasselbalch equation is derived: In this equation, αCO2 is the solubility coefficient of CO2 (0.03), and pK is the equilibrium constant for this buffer pair (6.1). Rearrangement yields the Henderson equation: Acid-base homeostasis depends on compensation for a primary disturbance. Compensation for a respiratory disturbance is metabolic, and compensation for a metabolic disturbance is respiratory. Furthermore, it is clear from the previous equations that in order to mitigate the change in proton concentration or pH, the direction of the compensation must be the same as the direction of the primary disturbance. Thus, consumption of bicarbonate will be accompanied by hyperventilation and a consequent reduction in PCO2. A simple acid-base disturbance is considered to consist of the primary disturbance and its normal compensation. A complex acid-base disturbance consists of more than one primary disturbance. In order to detect complex acid-base disturbances, one must be familiar with both the direction and magnitude of normal compensation (shown in Table 57.1).2 More than one metabolic disturbance may coexist (e.g., metabolic acidosis and metabolic alkalosis), but only one respiratory disturbance is possible at a time. In the present section, we will discuss disorders that affect the metabolic component of acid-base homeostasis: metabolic acidosis and metabolic alkalosis. Respiratory disturbances affecting acid-base balance will be discussed elsewhere (Chapters 37 and 40). An adult eating a normal diet generates 16,000 to 20,000 mmol of acid a day.3 Almost all of that acid is in the form of carbonic acid, resulting from CO2 and water generation in the metabolism of carbohydrates and fats. Individuals with normal ventilatory capacity eliminate this prodigious acid load through the lungs, thus the term volatile acid. The remainder of the daily acid load, about 1 mmol/kg body weight per day, derives from metabolism of phosphate- and sulfate-rich protein (yielding phosphoric and sulfuric acid). These nonvolatile or fixed acids are buffered, primarily by extracellular bicarbonate under normal circumstances. The kidneys are responsible for regenerating the consumed bicarbonate by secreting hydrogen ions (protons) in the distal nephron. These secreted protons must be buffered in the tubule lumen in order to allow elimination of the daily fixed acid load within the physiologic constraint of the minimum urinary pH. The urinary buffers are composed of the filtered sodium salts of the phosphoric acid and ammonia, which is synthesized in the proximal tubule and acidified in the collecting duct to form ammonium (NH4+). Under conditions of acid loading, the normal kidney reabsorbs all the filtered bicarbonate in the proximal tubule. Urinary net acid excretion therefore comprises phosphoric acid (so-called titratable acidity, because it is quantified by titrating the urine with alkali to pH 7.40) and ammonium, less any excreted bicarbonate.4 Many factors modify the kidney’s capacity to regulate acid-base balance. For example, renal ammoniagenesis is stimulated by acidemia, and inhibited by alkalemia, and thus participates in a homeostatic feedback loop.1 Hyperkalemia inhibits and hypokalemia stimulates renal ammoniagenesis. Hypokalemia further stimulates acid secretion by activating the Na+-H+ exchanger in the proximal tubule and the H+/K+-ATPase in the collecting duct. Finally, aldosterone stimulates both proton and K+ secretion in the collecting duct. For these reasons, hypokalemia tends to perpetuate a metabolic alkalosis, and hyperkalemia a metabolic acidosis.1 Metabolic acidosis can be caused by excessive production of fixed acid, decreased renal secretion of fixed acid, or loss of bicarbonate, either through the kidney or through the intestine.4 The net effect of any of these processes is a reduction in the blood bicarbonate concentration. The plasma anion gap helps to distinguish among the various causes of metabolic acidosis. Of course, because of charge neutrality, the sum of the concentration of all cations in the plasma is equal to the sum of all the anions. By convention, however, the anion gap is defined as the difference between the plasma sodium concentration and the sum of the bicarbonate and chloride concentrations. It represents the concentration of anions that are normally unmeasured by a basic metabolic chemistry panel.5 The anion gap normally is about 8 mmol/L, but it varies widely according to the methods employed by the clinical chemistry laboratory.6 The anion gap is composed mainly of albumin, along with phosphates, sulfates, and organic anions. There are two important pitfalls in the interpretation of the anion gap. First, because the anion gap is proportional to the plasma albumin concentration, hypoalbuminemia (common in critically ill patients) will lower the “baseline” anion gap (by approximately 2.5 mmol/L for each g/dL decline in the albumin concentration).7 Thus, profound hypoalbuminemia may falsely lower the anion gap, and thus mask a high anion gap acidosis. Second, alkalemia increases the anion gap by causing lactate generation and by titrating plasma buffers, most notably albumin.8 (Thus, in respiratory alkalosis, the bicarbonate concentration will be low in compensation, and the anion gap may be elevated, giving a false impression of a high anion gap metabolic acidosis by inspection of the electrolytes alone.) If bicarbonate is lost (e.g., through diarrhea), or hydrochloric acid is gained (e.g., renal tubular acidosis or administration of unbuffered amino acid solutions9), the bicarbonate concentration falls with a commensurate increase in the plasma chloride concentration; thus the anion gap is unchanged. If, on the other hand, bicarbonate is lost in buffering an organic acid such as lactic acid or a ketoacid, the decrement in the bicarbonate concentration is more or less matched by an increase in the anion gap. These processes are illustrated in Figure 57.1. Box 57.1 lists the causes of hyperchloremic metabolic acidosis. Two diagnoses are of particular interest in the critical care arena. First is the posthypocapnic metabolic acidosis, in which bicarbonate falls in compensation for a chronic respiratory alkalosis. When “normal” ventilation is restored, the pH falls until bicarbonate can be retained, giving the appearance of a hyperchloremic metabolic acidosis. This emphasizes the importance of observation over time in the analysis of acid-base status. The second entity of interest is a so-called dilutional hyperchloremic acidosis. This is seen in patients who are rapidly resuscitated with large volumes of isotonic saline solution. The acidosis traditionally has been attributed to dilution of blood bicarbonate. Analysis based on physical-chemistry principles may better explain the phenomenon (see later).10 The differential diagnosis of high anion gap metabolic acidosis is limited (Box 57.2). The most common cause in critically ill patients is a lactic acidosis. The causes of lactic acidosis are numerous. As shown in Box 57.3, they are divided into type A (imbalance between tissue oxygen demand and supply) and type B (impaired oxygen utilization).5 Diabetic ketoacidosis (DKA) (Chapter 58) and intoxications (Chapter 68) are discussed elsewhere. Two causes of high anion gap acidosis recently added to the differential diagnosis, and of particular relevance to intensivists, are pyroglutamic acidosis and intoxication with propylene glycol. Pyroglutamic acid is a metabolic intermediate in the γ-glutamyl cycle, one product of which is glutathione. Pyroglutamic acidosis may be congenital (caused by one of several enzyme deficiencies) or acquired.8 The acquired syndrome may be caused by acetaminophen (which depletes glutathione, leading to uninhibited pyroglutamic acid synthesis), β-lactam antibiotics, or glycine deficiency. The acidosis may be profound and the anion gap greater than 30 mmol/L.11 Definitive diagnosis is made by urinary screen for organic acids. In practice, however, circumstantial evidence suggests the acquired syndrome and the diagnosis is supported by a favorable response to appropriate intervention. Propylene glycol is a solvent for medications, many of which are commonly infused intravenously in critically ill patients, such as lorazepam, nitroglycerin, etomidate, and phenytoin. Propylene glycol is metabolized by alcohol dehydrogenase to lactic acid. High anion gap acidosis has been associated with high- and even low-dose infusions, particularly of lorazepam.7,12 Thus, development of a high anion gap acidosis in a critically ill patient should prompt a search for a source of propylene glycol, because withdrawal of the agent will promptly alleviate the acidosis. It has been generally accepted that severe acidemia (pH < 7.20) is associated with a variety of deleterious effects. Of particular concern are the cardiovascular effects, including pressure-resistant arterial vasodilation, venoconstriction, diminished myocardial contractility, and impaired hepatic and renal perfusion.13 (Some controversy exists as to which of these effects are directly caused by acidemia.4) A predisposition to malignant arrhythmias has been reported in vitro and in animal models. Finally, numerous metabolic derangements have been attributed to the effect of acidemia on key enzymes in metabolic pathways, resulting in sympathetic hyperactivity with diminished catecholamine responsiveness; insulin resistance and suppressed glycolysis; and reduced hepatic lactic acid uptake and metabolism.14 Acid-base disorders are revealed most commonly through the basic metabolic chemistry panel, when the plasma bicarbonate concentration is noted to be outside the normal range. If the bicarbonate is low, and if the anion gap is clearly elevated on that sample, a diagnosis of high anion gap metabolic acidosis can be made with some confidence, keeping in mind the pitfalls in the interpretation of the anion gap mentioned earlier.7 Once the primary disturbance has been identified, the astute clinician, recognizing the possibility of a mixed disturbance, is obligated to ask, “Is that all there is?” This question can be answered only by an understanding of the rules of normal compensation for simple acid-base disorders (see Table 57.1).2 Knowing at least the expected direction of compensation will allow the clinician to diagnose the most obvious mixed disturbances. For example, if the pH is low, the bicarbonate is low, and the PCO2 is above 40 mm Hg, there is clearly a mixed metabolic and respiratory acidosis. Similarly, if the pH is high, the bicarbonate is high, and the PCO2 is below 40 mm Hg, the diagnosis is a mixed respiratory and metabolic alkalosis. More subtle mixed disorders can be diagnosed only by understanding not only the expected direction, but the expected magnitude of compensation. This will allow one to conclude, for example, whether the hyperventilation in a patient with metabolic acidosis is appropriate (expected compensation), inadequate (a separate respiratory acidosis), or excessive (a separate respiratory alkalosis). The preceding method permits the diagnosis of simple and dual acid-base disorders. Triple acid-base disorders can be diagnosed only by comparing the change in the anion gap with the change in the plasma bicarbonate concentration. Most simply conceived, the fall in the bicarbonate should equal the rise in the anion gap (see Fig. 57.1). If the rise in the anion gap exceeds the fall in the bicarbonate, a metabolic alkalosis is said to be present in addition to the high anion gap acidosis. Conversely, if the fall in the bicarbonate exceeds the rise in the anion gap, mixed hyperchloremic and high anion gap acidoses are said to coexist. Although this analysis is useful in the case of large discrepancies, in more subtle cases it is confounded by theoretical and practical considerations.5,15 The classical approach to acid-base disorders described earlier has been challenged recently by proponents of a physical-chemistry approach described originally by Stewart.16 According to this method, the pH of the blood depends on the ionization of water by the difference in the concentration of so-called strong ions (the strong ion difference, or SID). The SID offers a quantitative approach to measuring the degree of acidosis in hyperchloremic metabolic acidosis. Although there is evidence that this approach may offer prognostic capabilities in patients with severe sepsis and septic shock with hyperchloremic metabolic acidosis, the complexity of the equations for calculating the SID may make this method cumbersome in clinical settings.17 The main utility of this construct in the critical care setting seems to be its explanation of a hyperchloremic metabolic acidosis in patients who receive large volumes of isotonic saline. The difficulty in accurately estimating this value arises from two factors: First, the apparent volume of distribution of bicarbonate varies more than twofold from 50% of body weight to 100% of body weight and is inversely proportional to the initial bicarbonate concentration.18 Second, there are often many simultaneous processes in a critically ill patient that tend to ameliorate or exacerbate the metabolic acidosis, such as vomiting, shock, and liver failure. In order to avoid overshoot alkalemia, it is prudent to estimate the volume of distribution to be 50% of the body weight14 and to target an increase in the bicarbonate concentration of no more than 8 mmol/L over 12 to 24 hours. Sodium bicarbonate generally is considered to be the alkalinizing agent of choice for severe acidemia. Alternative alkalinizing agents such as citrate, acetate, and lactate, which under normal circumstances are oxidized in the liver to bicarbonate, should not be used to treat acidemia in patients with suspected or confirmed hepatic impairment or circulatory compromise. The sodium bicarbonate should be administered as a continuous infusion, the concentration of which should be guided by the patient’s serum sodium concentration. Bolus injection of undiluted ampules of sodium bicarbonate (1000 mmol/L) should be used with great restraint and only in patients with the most severe acidemia because of the risk of hyperosmolality. Large volumes of any bicarbonate solution can lead to volume overload, a reduction in the ionized calcium concentration (see “Hypocalcemia”), and increased generation of CO2. This last effect will tend to cause a respiratory acidosis in patients with ventilatory insufficiency. Plasma electrolytes and blood gases must be monitored frequently to guide adjustments in the composition of the solution and its rate of infusion. Tris(hydroxymethyl)aminomethane, or THAM, is an amino alcohol that buffers without generating CO2. It has the advantage, therefore, of avoiding a superimposed respiratory acidosis. It has been used successfully in animals and humans with various metabolic acidoses.13,19 It is eliminated by the kidney, and thus should be used with caution in the setting of renal insufficiency. Risks include hyperkalemia, hypoglycemia, and hepatic necrosis (in neonates).7 The treatment of choice for lactic acidosis is reversal of the underlying cause of the acidosis (see Box 57.3). Pending resolution of the underlying disorder, however, the intensivist is often confronted with an unstable patient who is profoundly acidemic. Treatment at this stage is controversial.5,13 The debate has focused on the potentially deleterious effects of bicarbonate administration in lactic acidosis.20 In addition to the effects mentioned earlier, bicarbonate in animal models of lactic acidosis has been associated with increased lactate generation, reduction in intracellular pH, increased venous PCO2, and reduction in cardiac output. (This last effect correlates well with the reduction in ionized calcium concentration.13) Studies in humans likewise show no improvement in cardiac output, morbidity rate, or mortality rate with bicarbonate.13 Continuous venovenous hemodialysis (e.g., CVVHD) may be a promising tool for treating lactic acidosis, because it provides large amounts of bicarbonate without the risks of volume overload or hypocalcemia. There are several reported cases of successful treatment of metformin-associated lactic acidosis using continuous hemodialysis.21,22 Because of its superior short-term clearance compared with continuous renal replacement therapy, however, conventional hemodialysis remains the preferred treatment for metformin intoxication.23 Treatment of DKA and the acidoses associated with other intoxications are discussed in Chapters 58 and 68, respectively. Metabolic alkalosis is a process leading to accumulation of extracellular bicarbonate that, if unopposed, will result in an increase in the plasma pH (alkalemia). It can be caused either by a gain of bicarbonate or a loss of fixed acid from the ECF. The causes of metabolic alkalosis have been described.24 In its pure form, it is accompanied by hypoventilation (CO2 retention).2 From a pathophysiologic perspective, metabolic alkalosis is divided into those factors that generate the alkalosis and those factors that maintain or perpetuate it.24,25 Metabolic alkalosis is generated by addition of bicarbonate to the blood. This can occur either by loss of acid from the body or by addition of exogenous alkali. Loss of acid may be from the stomach (e.g., vomiting or nasogastric suction) or kidney. Renal acid loss is enhanced by a high rate of sodium delivery to the distal nephron, high circulating mineralocorticoid levels, potassium depletion, and high rates of ammoniagenesis. Because of the kidney’s prodigious ability to excrete bicarbonate, however, addition of bicarbonate to the blood is not sufficient to cause a sustained metabolic alkalosis. Some mechanism(s) to maintain the alkalosis must prevail. The most common mechanism contributing to the maintenance of metabolic alkalosis is volume depletion, either absolute or relative (e.g., congestive heart failure), which (1) reduces glomerular filtration, (2) enhances tubular bicarbonate reabsorption, and (3) causes secondary hyperaldosteronism, further enhancing urinary acidification. Another common perpetuating factor is potassium depletion, which stimulates proton secretion at several sites along the nephron.24–26 Most cases of clinically significant metabolic alkalosis are maintained by loss of chloride or potassium. Although total body sodium (and hence, volume) derangements are not directly responsible for the generation and maintenance of the metabolic alkalosis, potassium and chloride depletion are commonly seen in settings of volume depletion or excess. Therefore, from a clinical standpoint, it is useful to approach the patient with metabolic alkalosis centering on the history and physical examination, with special attention to the ECF volume status, followed by sequential analysis of blood chemistries.26 Causes of metabolic alkalosis are shown in Box 57.4. One entity unique to critically ill patients is posthypercapnic metabolic alkalosis. This syndrome is caused by abrupt treatment (usually with tracheal intubation and mechanical ventilation) of a chronic respiratory acidosis. The renal bicarbonate retention that compensated for the chronic respiratory acidosis persists (because of volume depletion) after restoration of a normal PCO2, resulting in the high pH and high plasma bicarbonate characteristic of metabolic alkalosis. The key to the diagnosis is the history and sequential analysis of blood chemistries.24 Alkalemia in critically ill patients is associated with increased mortality rate.27 Patients with combined metabolic and respiratory alkalosis have a higher mortality rate than those with respiratory alkalosis alone, and mortality rate in alkalemia is roughly proportional to the pH.27 Although no causal relationship between alkalemia and mortality rate has been established, the pathophysiology of alkalemia is far from benign.25 First, metabolic alkalosis suppresses ventilation, causing CO2 retention and relative hypoxemia.28 Second, alkalemia acutely increases hemoglobin’s oxygen affinity (Bohr effect). Third, respiratory alkalosis causes vasoconstriction, particularly in the cerebral circulation.25 All these processes tend to decrease tissue oxygen delivery.29 (Note that chronic alkalemia inhibits 2,3-diphosphoglycerate synthesis, allowing normalization of the oxyhemoglobin desaturation curve, mitigating tissue hypoxia to some extent.) These alterations in tissue oxygen delivery could be responsible at least in part for some of the clinical manifestations of metabolic alkalosis. Because alkalemia causes a decrease in ionized calcium concentration (see discussion under “Calcium Homeostasis”), many of the neuromuscular manifestations of metabolic alkalosis overlap with those of hypocalcemia, including paresthesias, tetany, and a predisposition to seizures.1 The acutely diminished tissue oxygen delivery to the brain may contribute to initial confusion and obtundation seen with metabolic alkalosis. Metabolic alkalosis often is accompanied by hypokalemia and hypomagnesemia. Thus, there is an association between alkalosis and arrythmias,24 but an independent effect of the alkalosis on cardiac arrythmogenesis has not been established. Increases in blood lactate concentration may occur in patients with metabolic alkalosis due to upregulation of phosphofructokinase and thus glycolysis, and because of tissue hypoxia (see earlier).8 With severe metabolic alkalosis (arterial pH above 7.55), the tissue hypoxia may be so marked that compensatory hypoventilation will be overridden by hypoxic drive, resulting in a normal to low arterial PCO2 and elevated blood lactate levels (so-called “lactic alkalosis”).30 Treatment of metabolic alkalosis entails correcting the factor(s) responsible for its maintenance and, if possible, correcting the factor that generated the alkalosis. Once the underlying diagnosis is clear (see Box 57.4), therapy is usually straightforward. If the metabolic alkalosis is maintained by chloride depletion and ECF volume contraction, the intravascular volume should be restored to normal, usually with intravenous isotonic saline.24,25 Potassium should be given, as KCl, to replace any deficits (see “Disorders of Potassium Homeostasis”), because potassium depletion perpetuates the metabolic alkalosis. If nasogastric suction cannot be stopped, acid loss can be reduced by the use of H2 blockers and proton pump inhibitors. Treating patients with metabolic alkalosis in the setting of volume overload and diminished effective circulating volume (e.g., congestive heart failure, hepatic cirrhosis) is more challenging, because saline infusion is contraindicated. Unless hyperkalemia is present, chloride should be replenished with KCl supplementation. In rare cases of concurent hyperkalemia, acetazolamide (a carbonic anydrase inhibitor) may be of benefit as it produces a bicarbonate diuresis. Acetazolamide should be avoided in patients with hypokalemia, because the alkaline diuresis will cause renal potassium wasting.24 Another potential complication of acetazolamide administration, particularly in patients with impending ventilatory failure, is worsening of hypercapnia owing to inhibition of red blood cell carbonic anhydrase and impaired CO2 transport.25 Hydrochloric acid infusion, as a 0.1 to 0.25 N solution, has been used with success in patients with severe metabolic alkalosis (pH > 7.55 and systemic instability such as encephalopathy or cardiac arrhythmia24) refractory to conventional measures.25,31 Correction of the metabolic disturbances has been reported with infusion of 0.25 N HCl at 100 mL/hour over about 12 hours.31 Extreme care must be taken to ensure that the infusion catheter is properly positioned within the vena cava, because the solution is highly caustic. Plasma chemistries must be monitored frequently in order to avoid overcorrection. If renal function is severely impaired or medical therapy is not possible, hemodialysis against a low-bicarbonate bath may be used.24 Disorders of potassium (K) homeostasis are common in hospitalized patients and may be associated with severe adverse clinical outcomes, including death.32,33 Prevention and proper treatment of hyper- and hypokalemia depend on an understanding of the underlying physiology. The total body potassium content of a 70-kg adult is about 3500 mmol, of which only 2% (about 70 mmol) is extracellular.34 This uneven distribution reflects the large potassium concentration gradient between the intracellular (Ki ≈140 mmol/L) and the extracellular (Ke ≈ 4.5 mmol/L) space, a gradient that is maintained by the intrinsic ion permeabilities of cell membranes and by Na+/K+-ATPase, the sodium-potassium pump.35 The Ke : Ki ratio largely determines the resting membrane potential of cells and thus is crucial for proper function of excitable tissues (muscle and nerve).35 Small absolute changes in Ke will perturb the ratio significantly. Therefore, disturbances of Ke (measured as changes in plasma potassium concentration, or PK) may have serious, even fatal, consequences mainly in the form of excitable tissue dysfunction. Internal potassium balance serves to protect against changes in Ke; potassium tends to move out of cells during potassium depletion and into cells following potassium intake. This process tends to prevent drastic alterations of Ke : Ki.36,37 The factors that influence internal potassium balance include hormones, acid-base status, plasma tonicity, exercise, and cell integrity (Box 57.5). The direction and magnitude of an acid-base-related change in PK depend on the nature and the duration of the disturbance. The most consistent and pronounced relationship between changes in pH and PK occurs in acute mineral (hyperchloremic) acidosis, where there is a strong inverse relationship between these two variables.38–40 Interestingly, hypokalemia is seen with prolonged mineral acidosis in patients with normal renal function and reflects increased renal potassium excretion.39 Unlike mineral acidoses, however, even severe acute organic (high anion gap) acidoses are not usually associated with hyperkalemia.41–44 Indeed, organic acidoses, such as lactic acidosis, actually tend to cause cellular potassium uptake.41 Nonetheless, factors coincident with the acidosis may alter PK. For example, mesenteric ischemia may result in both lactic acidosis (from anaerobic metabolism) and hyperkalemia. Even the hyperkalemia so commonly seen in patients with DKA does not result from the acidemia; rather, it appears to be a consequence of the characteristic insulin deficiency and hyperglycemia (see discussion of hypertonicity in the next paragraph).44 Respiratory disturbances typically alter PK less than metabolic disturbances. Alkaloses, respiratory or metabolic, have less effect on PK than their corresponding acidoses.38 Bicarbonate administration, which was once thought to reduce the PK by stimulating cellular potassium uptake,45 is now known to have very little if any immediate effect on internal potassium balance,46,47 except perhaps in patients with preexisting severe metabolic acidosis.41 It is clear, however, that longstanding alkalemia causes urinary potassium losses that may over time result in profound potassium depletion.48 Hypertonicity, as seen with hypertonic fluid administration49 or diabetic hyperglycemic states,50 leads to hyperkalemia, probably as a result of potassium efflux from cells by way of solvent drag. Fatal hyperkalemia has been attributed to this phenomenon in diabetic patients with end-stage renal disease (ESRD).51 Exercise causes a transient shift of potassium out of cells. Clinically significant hyperkalemia may result from exercise52,53 (and clinically misleading local venous hyperkalemia results from fist clenching during phlebotomy54). In contrast to the prodigious capacity of the kidney to excrete potassium,55 renal potassium conservation is imperfect and explains why significant potassium depletion and hypokalemia may result from dietary potassium deficiency alone.56 Normally, 90% to 95% of dietary potassium is eliminated through the kidney, and only about 5% to 10% through the intestine. It is the kidney that is almost entirely responsible for matching potassium output to potassium intake in order to maintain total body potassium constant.57 The majority of potassium excreted by the kidney derives from potassium secretion in the distal nephron (connecting tubule and collecting duct).57 Virtually all regulation of potassium excretion takes place at this site in the nephron, under the influence of two principal factors: the rate of flow and sodium delivery through that part of the nephron, and the effect of aldosterone.57 Potassium secretion is directly proportional to flow rate and sodium delivery through the distal nephron, explaining in part why diuretic use often is accompanied by hypokalemia.
Acid-Base, Electrolyte, and Metabolic Abnormalities
Acid-Base Homeostasis
Normal Acid-Base Physiology
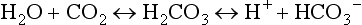
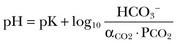
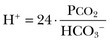
Metabolic Acidosis
Definition and Classification
Consequences of Acidemia
Diagnosis of Acid-Base Disorders
Treatment of Metabolic Acidosis
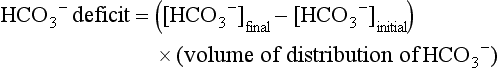
Metabolic Alkalosis
Definition and Classification
Clinical Consequences
Treatment
Potassium Homeostasis
Normal Potassium Physiology
Regulation of Internal Potassium Balance
Regulation of External Potassium Balance
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Acid-Base, Electrolyte, and Metabolic Abnormalities