FIGURE 55.1 Influence of 50% carboxyhemoglobin on the relationship between oxygen tension (PO2) and oxygen content.
Other Effects
Carbon monoxide has toxic effects that are unrelated to COHb (4). These effects include: (a) inhibition of cytochrome oxidase, which impairs the ability to generate ATP from oxidative metabolism, (b) release of nitric oxide from platelets, which promotes the formation of peroxynitrite, a powerful oxidant capable of widespread cell injury, and (c) activation of neutrophils, which is an additional source of oxidant injury (see Figure 14.1). These effects may play an important role in CO poisoning, because there is a poor correlation between COHb levels and the severity of carbon monoxide poisoning (1,4).
Clinical Features
The following statements summarize the clinical manifestations of CO poisoning, which are variable and nonspecific.
1. There is no correlation between the clinical manifestations of CO poisoning and COHb levels in blood (1,4).
2. Headache (usually frontal) and dizziness are the earliest and most common complaints in CO poisoning (reported in 85% and 90% of patients, respectively) (4).
3. Progressive exposure to CO can produce ataxia, confusion, delirium, generalized seizures, and coma (4).
4. Cardiac effects of CO poisoning include elevated biomarkers with normal coronary angiography, and transient LV systolic dysfunction (5).
5. Advanced cases of CO poisoning can be accompanied by rhabdomyolysis, lactic acidosis, and acute respiratory distress syndrome (ARDS) (4).
6. The “cherry red” skin color in classic descriptions of CO poisoning (because COHb is a brighter shade of red than hemoglobin) is a rare finding (1).
Delayed Neurologic Sequelae
Acute CO intoxication can be followed by the emergence (within about one year) of a variety neurologic abnormalities, mostly involving cognitive deficits (from mild confusion to severe dementia) and parkinsonism (1,4,6). These occur most frequently following prolonged (≥24 hrs) exposure to CO, and in patients with loss of consciousness or COHb levels above 25% (1). The mechanism for these delayed reactions has not been identified.
Diagnosis
The diagnosis of CO poisoning is not possible based on signs and symptoms alone. The diagnosis is suspected when the presenting signs and symptoms are consistent with CO poisoning, and the clinical history identifies a likely source of CO exposure (e.g., a house fire). The diagnosis is confirmed by an elevated COHb level in blood.
Carboxyhemoglobin
The measurement of hemoglobin in its different forms (oxygenated and deoxygenated hemoglobin, methemoglobin, and carboxyhemoglobin) is based on light absorption; this method is known as oximetry, and is described in Chapter 21. The following statements summarize the use of oximetry to measure COHb levels in blood.
1. Pulse oximetry is NOT reliable for the detection of COHb. Pulse oximeters use 2 wavelengths of light to measure oxygenated and deoxygenated hemoglobin in blood. Light absorbance at one of the wavelengths (660 nm) is very similar for oxygenated hemoglobin and COHb (see Figure 21.1 on page 410), so COHb is measured as oxygenated hemoglobin by pulse oximeters, and this results in spuriously high readings for O2 saturation (1).
2. The measurement of COHb requires an 8-wavelength oximeter (known as a CO-oximeter) that is available in most clinical laboratories. This device measures the relative abundance of all 4 forms of hemoglobin in blood.
COHb levels are negligible (<1%) in healthy nonsmokers, but smokers have COHb levels of 3–5% or even higher (1). The threshold for elevated COHb levels is 3–4% for nonsmokers, and 10% for smokers (1).
Treatment
The treatment for CO poisoning is inhalation of 100% oxygen. The elimination half-life of COHb is 320 minutes while breathing room air, and 74 minutes while breathing 100% oxygen (1), so only a few hours of breathing pure oxygen is needed to return COHb levels to normal.
Hyperbaric Oxygen
Hyperbaric oxygen has been advocated as a means of reducing the delayed neurologic sequelae in CO poisoning, but evidence of benefit has not been convincing (7).
Cyanide
House fires generate cyanide as well as carbon monoxide, and the possibility of cyanide poisoning must be considered in all patients with CO poisoning from a house fire. When smoke inhalation from a house fire is accompanied by a severe metabolic acidosis (pH <7.2) or a markedly elevated serum lactate level (≥10 mmol/L), empiric treatment for cyanide poisoning is recommended (1). See the next section for the treatment of cyanide poisoning.
CYANIDE
Cyanide is a lethal toxin with a nefarious history. The Nazis used hydrogen cyanide gas (Zyklon-B) for mass murder in the 1940s, and in 1978, cyanide-laced fruit drinks were used for mass murder/suicide by Jim Jones and his followers. The principal source of cyanide poisoning is inhalation of hydrogen cyanide gas during domestic fires (8,9). Infusion of the vasodilator, sodium nitroprusside, is an additional source of cyanide toxicity in ICU patients (see Figure 53.4).
Pathophysiology
Cyanide ions have a high affinity for metalloproteins, including the oxidized iron (Fe3+) in cytochrome oxidase and methemoglobin, and the cobalt in hydroxocobalamin (a precursor of vitamin B12). Cytochrome oxidase is situated at the end of the electron-transport chain in mitochondria, and is the site where the electrons collected during ATP production are used to reduce oxygen to water (thereby clearing the way for continued ATP production). Cyanide-induced inhibition of cytochrome oxidase halts the process of oxidative metabolism in mitochondria. This halts the uptake of pyruvate into mitochondria, and results in excess production of lactic acid and a progressive metabolic (lactic) acidosis. The defect in cellular energy production is fatal if not corrected.
Cyanide Clearance
There are two endogenous mechanisms for clearing cyanide from the body, and these are shown in Figure 53.4 (see page 953). The principal mechanism is the transfer of sulfur from thiosulfate (S2O3) to cyanide, which forms thiocyanate (SCN). This is called a transulfuration reaction.
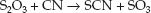 (55.1)
(55.1)
Thiocyanate is then cleared by the kidneys. Thiocyanate can accumulate in patients with renal failure, and can cause an acute psychosis (10).
The second (minor) mechanism for cyanide clearance is the reaction of cyanide with methemoglobin to form cyanomethemoglobin; i.e.,
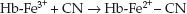 (55.2)
(55.2)
The hemoglobin-bound cyanide is eventually cleared via transulfuration. These two clearance mechanisms are easily overwhelmed, especially in the setting of thiosulfate deficiency (e.g., in smokers).
Clinical Features
Early signs of cyanide poisoning include agitation, tachycardia, and tachypnea, representing the compensatory stage of metabolic acidosis. Progressive cyanide accumulation eventually results in loss of consciousness, bradycardia, hypotension, and cardiac arrest. Plasma lactate levels are typically very high (>10 mmol/L), and venous blood can look “arterialized” because of the marked decrease in tissue O2 utilization. Pro-gression is rapid after smoke inhalation, and the time from onset of symptoms to cardiac arrest can be less than 5 minutes (8).
Diagnosis
Cyanide poisoning is a clinical diagnosis. Whole blood cyanide levels can be used for documentation, but results are not immediately available, and cyanide antidotes must be given quickly for optimal results. The clinical diagnosis of cyanide poisoning is particularly challenging in victims of smoke inhalation, because many of the clinical features of cyanide poisoning are indistinguishable from carbon monoxide (CO) poisoning. As a general rule of thumb, severe metabolic (lactic) acidosis and hemodynamic instability are the clinical features that distinguish cyanide poisoning from CO poisoning in victims of smoke inhalation (8,9).
Treatment
Antidote therapy should begin immediately after cyanide poisoning is first suspected. The antidotes for cyanide poisoning are presented in Table 55.1.
Hydroxocobalamin
The antidote of choice for cyanide poisoning is hydroxocobalamin, a cobalt-containing precursor of vitamin B12 that combines with cyanide to form cyanocobalamin, which is then excreted in the urine. The recommended dose is 5 grams, as an IV bolus. A second dose of 5 grams is recommended for patients with cardiac arrest (8). Hydroxocobalamin is relatively safe to use, but urine and other body fluids can have a reddish color for a few days.
Table 55.1 Antidotes for Cyanide Poisoning
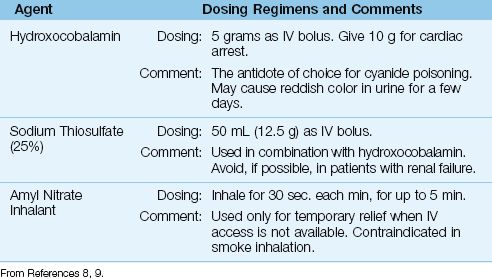
Sodium Thiosulfate
Sodium thiosulfate converts cyanide to thiocyanate (see Equation 55.1), and is used in combination with hydroxocobalamin. The recommended dose is 12.5 grams as an IV bolus. Since thiocyanate can accumulate in renal failure and cause an acute psychosis (10), thiosulfate should not be used in patients with renal failure. If thiosulfate is given before evidence of renal failure is available, watch for signs of thiocyanate toxicity (which is treated with hemodialysis).
Nitrates
Nitrates promote cyanide clearance by promoting the formation of methemoglobin. However, nitrates have undesirable side effects, and are contraindicated in smoke inhalation (because they cause a leftward shift in the oxyhemoglobin dissociation curve, and can aggravate the similar effect of carbon monoxide). The only role for nitrates in cyanide poisoning is the use of inhaled amyl nitrate as a temporary measure when IV access is not available.
Cyanide Antidote Kits
There are specialized antidote kits for cyanide poisoning (e.g., Akorn Cyanide Antidote Kit) that contain amyl nitrate for inhalation, sodium nitrite (300 mg in 10 mL) for IV injection, and sodium thiosulfate (12.5 g in 50 mL) for IV injection. These kits can be used as a source of thiosulfate, but they do not contain hydroxocobalamin (at least at the present time).
TOXIC ALCOHOLS
Ethylene glycol and methanol are common components of household, automotive, and industrial products, and they both produce toxidromes characterized by a metabolic acidosis. They are known as toxic alcohols (10), which is a misnomer, because it implies that ethanol is non-toxic. Toxic alcohols are the 12th leading source of toxic exposures in the United States (11).
Ethylene Glycol
Ethylene glycol is the main ingredient in many automotive antifreeze products. It has a sweet, agreeable taste, which makes it a popular method of attempted suicide.
Pathophysiology
Ethylene glycol is readily absorbed from the GI tract, and 80% of the ingested dose is metabolized in the liver. As shown in Figure 55.2, the metabolism of ethylene glycol involves the formation of a series of acids, with the participation of alcohol dehydrogenase and lactate dehydrogenase enzymes, and ends with the formation of oxalic acid (12). Each of the intermediate reactions involves the conversion of NAD to NADH, which promotes the conversion of pyruvate to lactate. As a result, serum lactate levels are also elevated in ethylene glycol poisoning (12). Each of the acid intermediates in ethylene glycol metabolism is a strong acid that readily dissociates, and can contribute to a metabolic acidosis. The oxalic acid also combines with calcium to form insoluble calcium oxalate crystals that precipitate in several tissues, and are particularly prominent in the renal tubules. These crystals can be a source of renal tubular injury.
Clinical Features
Early signs of ethylene glycol intoxication include nausea, vomiting, and apparent inebriation (altered mental status, slurred speech, and ataxia). Because ethylene glycol is odorless, there is no odor of alcohol on the breath. Severe cases are accompanied by depressed consciousness, coma, generalized seizures, renal failure, pulmonary edema, and cardiovascular collapse (12). Renal failure can be a late finding (24 hours after ingestion).
Laboratory studies show a metabolic acidosis with an elevated anion gap and an elevated osmolal gap (see page 656 for a description of the osmolal gap). Serum lactate levels can be elevated (usually 5–6 mEq/L). A plasma assay for ethylene glycol is available, and a level >20 mg/dL is considered toxic, but the results are not immediately available, and are not used in the decision to initiate therapy (12). (Plasma levels can be used to guide decisions about discontinuing therapy, as described later.)
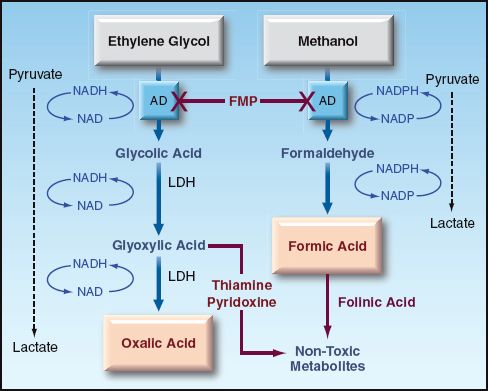
FIGURE 55.2 The metabolism of ethylene glycol and methanol in the liver. AD = alcohol dehydrogenase; LDH = lactate dehydrogenase; FMP = fomepizole.
CRYSTALLURIA: Calcium Oxalate Crystals Can Be Visualized In The Urine In About 50% Of Cases Of Ethylene Glycol Poisoning (14). The Presence Of Calcium Oxalate Crystals Is Not Specific For Ethylene Glycol Poisoning, But The Shape Of The Crystals Is More Specific; I.E., Thin, Monohydrate Crystals, Like The Ones Shown In Figure 55.3, Are More Characteristic Of Ethylene Glycol Poisoning Than The Box-Shaped Dihydrate Crystals (12). Most Hospital Laboratories Do Not Routinely Inspect Urine For Crystals, So Make Sure To Request A Search For Crystalluria When Ethylene Glycol Toxicity Is Supected.
Treatment
The management of ethylene glycol poisoning involves measures to alter the metabolism of ethylene glycol, along with hemodialysis, if necessary.
FOMEPIZOLE: Fomepizole Inhibits Alcohol Dehydrogenase, The Enzyme Involved In The Initial Step Of Ethylene Glycol Metabolism (See Figure 55.2). The Recommended Dosing Regimen For Both Ethylene Glycol And Methanol Poisoning Is Shown In Table 55.2. The Best Results Are Obtained If Therapy Begins Within 4 Hours Of Ingestion. Fomepizole Should Be Continued Until The Metabolic Acidosis Has Resolved, And The Plasma Level Of Ethylene Glycol Is In The Non-Toxic Range (12).

FIGURE 55.3 Microscopic appearance of calcium oxalate monohydrate crystals. The presence of these thin, needle-shaped crystals in the urine is highly suggestive of ethylene glycol poisoning.
Table 55.2Dosing Regimen for Fomepizole

HEMODIALYSIS: The clearance of ethylene glycol and all its metabolites is enhanced by hemodialysis. The indications for immediate hemodialysis include severe acidemia (pH <7.1), and evidence of significant end-organ damage (e.g., coma, seizures, and renal insufficiency) (12). Multiple courses of hemodialysis may be necessary, and fomepizole dosing should be adjusted, if hemodialysis is continued, as indicated in Table 55.2.
ADJUNCTS: THIAMINE (100 MG IV DAILY) AND PYRIDOXINE (100 MG IV DAILY) ARE RECOMMENDED TO DIVERT GLYOXYLIC ACID TO THE FORMATION OF NON-TOXIC METABOLITES (SEE Figure 55.2).
Methanol
Methanol (also known as wood alcohol because it was first distilled from wood) is a common ingredient in shellac, varnish, windshield washer fluid, and solid cooking fuel (Sterno) (12).
Pathophysiology
Like ethylene glycol, methanol is readily absorbed from the upper GI tract, and is metabolized by alcohol dehydrogenase in the liver. The principal metabolite is formic acid, a strong acid that readily dissociates and produces a metabolic acidosis with a high anion gap. Formic acid is also a mitochondrial toxin that inhibits cytochrome oxidase and blocks oxidative energy production. Tissues that are particularly susceptible to damage are the retina, optic nerve, and basal ganglia (12). Methanol metabolism promotes the conversion of pyruvate to lactate in the same manner described for ethylene glycol metabolism, and lactate production is increased further by the actions of formic acid on cytochrome oxidase activity.
Clinical Features
Early manifestations (within 6 hours of ingestion) include signs of apparent inebriation without the odor of alcohol on the breath (as in ethylene glycol intoxication). Later signs (6–24 hours after ingestion) include visual disturbances (e.g., scotoma, blurred vision, complete blindness), depressed consciousness, coma, and generalized seizures (12). Examination of the retina can reveal papilledema and generalized retinal edema. Visual disturbances are characteristic of methanol poisoning, and are not a feature of ethylene glycol poisoning.
Laboratory studies show a high-anion-gap metabolic acidosis and an elevated osmolal gap, similar to ethylene glycol poisoning. However, there is no crystalluria in methanol poisoning. A plasma assay for methanol is available, and a level above 20 mg/dL is considered toxic. However, the re-sults of the plasma assay are not immediately available, and are not used in the decision to initiate therapy.
Treatment
Treatment for methanol poisoning is the same as described for ethylene glycol poisoning, except for the following: (a) visual impairment is an indication for dialysis in methanol poisoning, and (b) folinic acid is used as adjunctive therapy in methanol poisoning, instead of thiamine and pyridoxine.
OLINIC ACID: Folinic Acid (Leucovorin) Can Convert Formic Acid To Non-Toxic Metabolites. The Recommended Dose Is 1 Mg/Kg Iv, Up To 50 Mg, At 4-Hour Intervals (12). Folic Acid Should Be Used If Folinic Acid Is Unavailable.
A FINAL WORD
The following points in this chapter deserve emphasis:
1. Carbon monoxide poisoning can be missed if you rely on pulse oximetry readings to detect O2 desaturation. Detection of elevated carboxyhemoglobin levels requires an 8-wavelength CO-oximeter, which is available in most clinical laboratories.
2. A victim of smoke inhalation who has a severe metabolic acidosis should be treated empirically for cyanide poisoning.
3. Methanol and ethylene glycol poisoning should be considered in any patient who presents with a high-anion-gap metabolic acidosis of unclear etiology.
4. If ethylene glycol poisoning is a consideration, inspection of the urine for calcium oxalate monohydrate crystals can provide useful information.
REFERENCES
Carbon Monoxide
1. Hampson NB, Piantadosi CA, Thom SR, Weaver LK. Practice recommendations in the diagnosis, management, and prevention of carbon monoxide poisoning. Am J Resp Crit Care Med 2012; 186:1095–1101.
2. Guyton AC, Hall JE. Medical Physiology, 10th ed. Philadelphia: W.B. Saunders, Co, 2000:470.
3. Nunn JF. Nunn’s Applied Respiratory Physiology. 4th ed. Oxford: Butterworth Heinemann, 1993:279–280.
4. Guzman JA. Carbon monoxide poisoning. Crit Care Clin 2012; 28:537–548.
5. Kalay N, Ozdogru I, Cetinkaya Y, et al. Cardiovascular effects of carbon monoxide poisoning. Am J Cardiol 2007; 99:322–324.
6. Choi IS. Delayed neurologic sequelae in carbon monoxide intoxication. Arch Neurol 1983; 40:433–435.
7. Buckley NA, Juurlick DN, Isbister G, et al. Hyperbaric oxygen for carbon monoxide poisoning. Cochrana Database Syst Rev 2011; 4:CD002041.
Cyanide
8. Anseeuw K, Delvau N, Burill-Putze G, et al. Cyanide poisoning by fire smoke inhalation: a European expert consensus. Eur J Emerg Med 2013; 20:2–9.
9. Baud FJ. Cyanide: critical issues in diagnosis and treatment. Hum Exp Toxicol 2007; 26:191–201.
Toxic Alcohols
10. Weiner SW. Toxic alcohols. In Nelson LS, Lewin NA, Howland MA, et al., eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York: McGraw-Hill, 2006:1400–1410.
11. Bronstein AC, Spyker DA, Cantilena LR, Jr, et al. 2011 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 29th Annual Report. Clin Toxicol 2012; 50:911–1164.
12. Kruse PA. Methanol and ethylene glycol intoxication. Crit Care Clin 2012; 28:661–711.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


