1754
Calcium and phosphorus are responsible for much of the structural integrity of the bony skeleton. Although neither is found in abundance in the soft tissues, both have an important role in vital cell functions. Phosphorus participates in aerobic energy production, whereas calcium participates in blood coagulation, neuromuscular transmission, and smooth muscle contraction. Considering the important functions of these electrolytes, it is surprising that abnormalities in calcium and phosphorus balance are so well tolerated.
CALCIUM IN PLASMA
Calcium is the most abundant electrolyte in the human body (the average adult has more than half a kilogram of calcium), but 99% is in bone (1,2). In the soft tissues, calcium is 10,000 times more concentrated than in the extracellular fluids (2,3).
Plasma Calcium
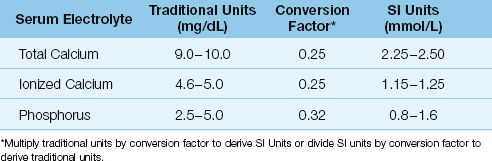
The calcium in plasma is present in three forms, as depicted in Figure 38.1. Approximately half of the calcium is ionized (biologically active) and the remainder is complexed (biologically inactive) (1). About 80% of the bound calcium is bound to albumin, while 20% is complexed to plasma anions such as proteins and sulfates. The concentration of total and ionized calcium in plasma is shown in Table 38.1. These values may vary slightly in different clinical laboratories.
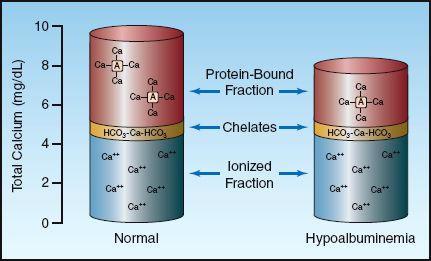
FIGURE 38.1 The three fractions of calcium in plasma and the contribution of each to the total calcium concentration. The column on the right shows how a decrease in plasma albumin can reduce the total plasma calcium without affecting the ionized calcium.
Total vs. Ionized Calcium
The calcium assay used by most clinical laboratories measures all three fractions of calcium, which can be misleading. This is demonstrated in the column on the right in Figure 38.1, which shows that a decrease in the plasma albumin concentration will decrease the total calcium concentration in plasma without affecting the ionized (biologically active) calcium concentration.
A variety of correction factors have been proposed for adjusting the plasma calcium concentration in patients with hypoalbuminemia. However, none of these correction factors are reliable (4,5), and the only reliable method of determining the concentration of ionized calcium is to measure it with ion-specific electrodes.
Table 38.1 Normal Ranges for Calcium and Phosphate in Blood
Ionized Calcium Measurement
Ionized calcium can be measured in whole blood, plasma, or serum with ion-specific electrodes that are now available in most clinical laboratories.
BLOOD COLLECTION Some precautions are necessary when collecting blood samples for an ionized calcium measurement (7). Loss of carbon dioxide from a blood sample could falsely lower the ionized calcium (by increasing calcium binding to albumin), so it is important to avoid gas bubbles in the blood sample. Anticoagulants (e.g., heparin, citrate, and EDTA) can bind calcium, so blood samples should not be placed in collection tubes that contain these anticoagulants. Tubes with red stoppers (“red top” tubes) contain silicone, and are adequate for measuring ionized calcium in serum samples.
IONIZED HYPOCALCEMIA
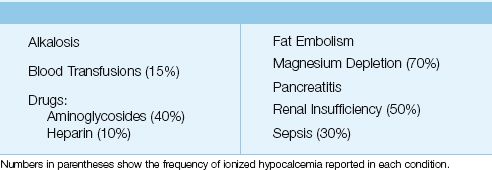
In a large, multicenter survey that included over 7,000 ICU patients, 88% of the patients had at least one episode of mild ionized hypocalcemia (0.9–1.14 mmol/L), and 3.3% of patients had at least one episode of severe ionized hypocalcemia (<0.8 mmol/L) (7). The common disorders associated with ionized hypocalcemia in ICU patients are listed in Table 38.2. Hypoparathyroidism is a leading cause of hypocalcemia in outpatients, but is not a consideration in the ICU unless a patient has had recent neck surgery.
Table 38.2 Causes of Ionized Hypocalcemia in the ICU
Predisposing Conditions
Magnesium Depletion
Magnesium depletion promotes hypocalcemia by inhibiting parathormone secretion and reducing end-organ responsiveness to parathormone (see Chapter 37 for references). Hypocalcemia from magnesium depletion is refractory to calcium replacement therapy, and magnesium replacement often corrects the hypocalcemia.
Sepsis
Sepsis is a common cause of hypocalcemia in the ICU (8,9), but the mechanism is unclear. Hypocalcemia is independent of the systemic vasodilation that accompanies sepsis (9), so the clinical significance of the hypo-calcemia is unclear.
Alkalosis
Alkalosis promotes the binding of calcium to albumin, which reduces the concentration of ionized calcium in blood. Symptomatic hypocalcemia is more common with respiratory alkalosis than with metabolic alkalosis. Infusions of sodium bicarbonate can also produce ionized hypocalcemia because calcium directly binds to the infused bicarbonate.
Blood Transfusions
Ionized hypocalcemia has been reported in 20% of patients receiving blood transfusions (8). The mechanism is calcium binding by the citrate anticoagulant in banked blood. Hypocalcemia from blood transfusions is usually transient, and resolves when the infused citrate is metabolized by the liver and kidneys (8). In patients with renal or hepatic failure, a more prolonged hypocalcemia can result. The hypocalcemia associated with blood transfusions does not impede blood coagulation; as a result, calcium infusions are not necessary in massive blood transfusions.
Drugs
A number of drugs can bind calcium and promote ionized hypocalcemia (8). The ones most often used in the ICU are aminoglycosides and heparin.
Renal Failure
Ionized hypocalcemia can accompany renal failure as a result of phosphate retention and impaired conversion of vitamin D to its active form in the kidneys. The treatment is aimed at lowering the phosphate levels in blood with antacids that block phosphorus absorption in the small bowel. However, the value of this practice is unproven. The acidosis in renal failure can decrease the binding of calcium to albumin, so hypocalcemia in renal failure does not imply ionized hypocalcemia.
Pancreatitis
Severe pancreatitis can produce ionized hypocalcemia through several mechanisms. The prognosis is adversely affected by the appearance of hypocalcemia (10), although a causal relationship between hypocalcemia and fatal outcomes has not been proven.
Clinical Manifestations
The major consequences of hypocalcemia are enhanced cardiac and neuromuscular excitability, and reduced contractile force in cardiac muscle and vascular smooth muscle. However, most cases of ionized hypocalcemia in the ICU have no apparent adverse consequences (7).
Neuromuscular
Hypocalcemia can be accompanied by tetany (of peripheral or laryngeal muscles), hyperreflexia, paresthesias, and seizures (11). Chvostek’s and Trousseau’s signs are often listed as manifestations of hypocalcemia, but Chvostek’s sign is nonspecific (it is present in 25% of normal adults), and Trousseau’s sign is insensitive (it can be absent in at least 30% of patients with hypocalcemia) (12).
Cardiovascular
The cardiovascular complications of hypocalcemia include hypotension, decreased cardiac output, and ventricular ectopic activity. However, these complications are reported only in cases of extreme ionized hypocalcemia (<0.65 mmol/L) (8).
Calcium Replacement Therapy
The treatment of ionized hypocalcemia should be directed at the underlying cause of the problem. If calcium replacement is needed, the intravenous calcium solutions and a recommended dosing regimen are shown in Table 38.3.
Table 38.3 Intravenous Calcium Replacement Therapy

Calcium Salt Solutions
The calcium solutions available for intravenous use are 10% calcium chloride and 10% calcium gluconate. Calcium chloride contains three times more elemental calcium than calcium gluconate, but calcium gluconate is usually preferred because it has a lower osmolarity, and is less irritating when injected. However, both calcium solutions are hyperosmolar, and should be given via a large central vein, if possible.
Dosing Regimen
A bolus dose of 200 mg elemental calcium (diluted in 100 mL isotonic saline and given over 5–10 minutes) should raise the total serum calcium by 0.5 mg/dL, but levels will begin to fall after 30 minutes (8). Therefore, the bolus dose of calcium should be followed by a continuous infusion at a dose rate of 1–2 mg/kg/hr (elemental calcium) for at least 6 hours. Individual responses will vary, so calcium dosing should be guided by the level of ionized calcium in blood (8).
CAUTION: Intravenous calcium can be risky in select patient populations. Calcium infusions can promote vasoconstriction and ischemia in any of the vital organs (13). The risk of calcium-induced ischemia should be particularly high in patients with low cardiac output who are already vasoconstricted. In addition, aggressive calcium replacement can promote intracellular calcium overload, which can produce lethal cell injury (14), particularly in patients with circulatory shock. Because of these risks, it is wise to avoid correcting ionized hypocalcemia with intravenous calcium unless there is evidence of a serious complication linked to the hypocalcemia.
Maintenance Therapy
The daily maintenance dose of calcium is 2–4 g in adults. This can be administered orally using calcium carbonate (e.g., Oscal) or calcium gluconate tablets (500 mg calcium per tablet).
IONIZED HYPERCALCEMIA
In the large survey of ionized calcium levels mentioned previously (7), 23% of ICU patients had at least one episode of mild ionized hypercalcemia (1.26–1.35 mmol/L), and 17% of patients had at least one episode of ionized hypercalcemia and ionized hypocalcemia. The source of ionized hypercalcemia in ICU patients has not been adequately studied, but common causes of hypercalcemia outside the ICU are hyperparathyroidism and malignancy (15–17).
Clinical Manifestations
The manifestations of hypercalcemia are nonspecific, and can be categorized as follows (16):
1. Gastrointestinal: nausea, vomiting, constipation, ileus, and pancreatitis
2. Cardiovascular: hypovolemia, hypotension, and shortened QT interval
3. Renal: polyuria and nephrocalcinosis
4. Neurologic: confusion and depressed consciousness, including coma
These manifestations can become evident when the total serum calcium is >12 mg/dL (or the ionized calcium is >3.0 mmol/L), and they are almost always present when the serum calcium is >14 mg/dL (or the ionized calcium is >3.5 mmol/L) (17). Manifestations are more likely with rapid rises in serum calcium.
Management
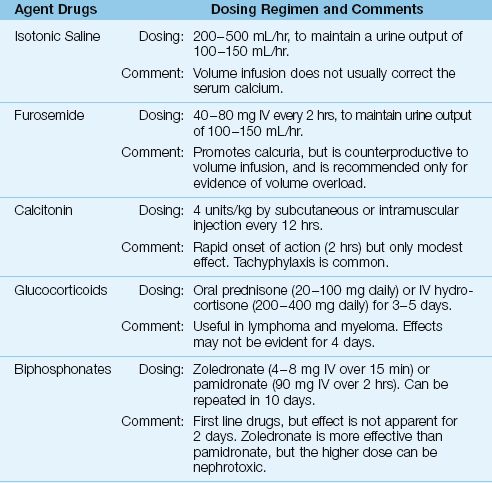
Treatment is indicated when the hypercalcemia is associated with adverse effects, or when the serum calcium is greater than 14 mg/dL (or ionized calcium >3.5 mmol/L). Most cases of severe, symptomatic hypercalcemia (hypercalcemic crisis) are cancer-related, and the treatment is summarized in Table 38.4 (1,15–17).
Table 38.4 Management of Severe Hypercalcemia
Isotonic Saline Infusion
Hypercalcemia usually is accompanied by hypercalciuria, which produces an osmotic diuresis. This eventually leads to hypovolemia, which reduces calcium excretion in the urine and precipitates a rapid rise in the serum calcium. Therefore, volume infusion to correct hypovolemia and promote renal calcium excretion is the first goal of management for hypercalcemia. Isotonic saline (200–500 mL/hr) is recommended for the volume infusion (16) because natriuresis promotes renal calcium excretion. The goal is a urine output of 100–150 mL/hr (15–17).
Furosemide
Saline infusion does not correct hypercalcemia in over 70% of cases (15), and furosemide is often recommended (40 to 80 mg IV every 2 hours) to further promote urinary calcium excretion. However, this can be counterproductive because it promotes hypovolemia, so furosemide is recommended only in cases of volume overload (15,17).
Calcitonin
Calcitonin is a naturally occurring hormone that inhibits bone resorption. It is available as salmon calcitonin, which is given subcutaneously or intramuscularly in a dose of 4 U/kg every 12 hours. The response is rapid (onset within a few hours), but the effect is mild (the maximum drop in serum calcium is 0.5 mmol/L) and tachyphylaxis is common (15). As a result, calcitonin has largely been abandoned for the treatment of severe hypercalcemia (15).
Glucocorticoids
Glucocorticoids have several effects that can decrease the serum calcium, including increased renal excretion of calcium, decreased osteoclast activity in bone, and decreased extrarenal production of calcitriol in lymphoma and myeloma (15). Glucocorticoid regimens include oral prednisone (40–100 mg daily) or intravenous hydrocortisone (200–400 mg daily) for 3–5 days. On the negative side, glucocorticoid effects may not be evident for 4 days, and they can precipitate tumor lysis syndrome (15).
Bisphosphonates
The biphosphonates are potent inhibitors of osteoclast activity. Two drugs in this class, zoledronate (4 or 8 mg IV over 15 min) and pamidronate (90 mg IV over 2 hours) are considered first line agents in the treatment of severe hypercalcemia (15). Zoledronate is the more effective agent, but the higher dose carries a risk of renal injury. Both drugs have a delayed onset of action (2–4 days). The peak effect is at 4–7 days, and the effect lasts 1–4 weeks (15).
Dialysis
Dialysis (hemodialysis or peritoneal dialysis) is effective in removing calcium in patients with renal failure.
HYPOPHOSPHATEMIA
Unlike calcium, inorganic phosphate (PO4) is predominantly intracellular in location, where it participates in glycolysis and high energy phosphate (ATP) production. The normal concentration of PO4 in plasma is shown in Table 38.1 (18).
Hypophosphatemia (serum PO4 <2.5 mg/dL or <0.8 mmol/L) is reported in 17–28% of critically ill patients (19,20), and can be the result of an intracellular shift of phosphorus, an increase in the renal excretion of phosphorus, or a decrease in phosphorus absorption from the GI tract. Most cases of hypophosphatemia are due to movement of PO4 into cells.
Predisposing Conditions
Glucose Loading
The movement of glucose into cells is accompanied by a similar movement of PO4 into cells, and this can lead to hypophosphatemia if extracellular PO4 levels are marginal. Glucose loading is the most common cause of hypophosphatemia in hospitalized patients (19,21,22), and typically occurs during refeeding in alcoholic, malnourished, or debilitated patients. The influence of parenteral nutrition on serum PO4 levels is shown in Figure 38.2. Note the gradual decline in the serum PO4 and the severe degree of hypophosphatemia (serum PO4 <1 mg/dL) after 7 days of intravenous nutrition. The risk of hypophosphatemia is one of the reasons why parenteral nutrition regimens are advanced gradually for the first few days.
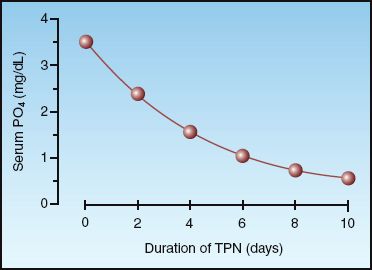
FIGURE 38.2 The cumulative effect of total parenteral nutrition (TPN) on the serum phosphate level. Data from Reference 21.
Prolonged Hyperglycemia
A phenomenon similar to glucose loading is seen in patients with prolonged hyperglycemia who receive insulin. The hyperglycemia produces an osmotic diuresis (from glycosuria), which promotes urinary PO4 losses, but the hypophosphatemia appears only when insulin is given (which drives PO4 into cells). An example of this phenomenon is the hypophosphatemia that appears during the treatment of diabetic ketoacidosis (see page 613).
Respiratory Alkalosis
Respiratory alkalosis can increase intracellular pH, and this accelerates glycolysis. The increase in glucose utilization is accompanied by an increase in glucose and phosphorus movement into cells (23). This may be an important source of hypophosphatemia in ventilator-dependent patients because overventilation and respiratory alkalosis are common in these patients.
ß-Receptor Agonists
Stimulation of α-adrenergic receptors can move PO4 into cells and promote hypophosphatemia. This effect is evident in patients treated with β-agonist bronchodilators. In one study of patients with acute asthma who were treated aggressively with nebulized albuterol (2.5 mg every 30 minutes), the serum PO4 decreased by 1.25 mg/dL (0.4 mmol/L) 3 hours after the onset of therapy (24).
Systemic Inflammation
There is an inverse relationship between serum PO4 and circulating levels of inflammatory cytokines (25). Possible explanations include in-creased PO4 utilization by activated neutrophils, and a transcellular shift of PO4 caused by elevated levels of endogenous catecholamines.
Phosphate Binding Agents
Aluminum can form insoluble complexes with inorganic phosphates. As a result, aluminum-containing compounds such as sucralfate (Carafate) can impede the absorption of phosphate in the upper GI tract and promote phosphate depletion (26).
Clinical Manifestations
Hypophosphatemia is often clinically silent, even when the serum PO4 falls to extremely low levels. In one study of patients with severe hypophosphatemia (i.e., serum PO4 <1 mg/dL), none of the patients showed evidence of harm (27). Despite the apparent lack of harm, phosphate depletion creates a risk for impaired energy metabolism.
Energy Metabolism
Phosphate depletion has several effects that could impair energy metabolism. These are summarized below, and indicated in Figure 38.3.
1. Cardiac Output: Phosphate depletion can impair myocardial contractility and reduce cardiac output. Hypophosphatemic patients with heart failure have shown improved cardiac performance after phosphate supplementation (28).
2. Erythrocytes: Reduction of high energy phosphate production from glycolysis in erythrocytes can reduce the deformability of red cells. This may explain why severe hypophosphatemia can be accompanied by a hemolytic anemia (26).
3. Oxyhemoglobin Dissociation: Phosphate depletion is accompanied by depletion of 2,3-diphosphoglycerate, and this shifts the oxyhemoglobin dissociation curve to the left. When this occurs, hemoglobin is less likely to release oxygen to the tissues.
4. Energy Availability: Phosphate depletion can reduce the availability of cellular energy by impeding the production of high-energy phosphate compounds (ATP).
Muscle Weakness
There is one report of respiratory muscle weakness and failure to wean from mechanical ventilation in patients with severe hypophosphatemia (29). However, other studies have not shown significant respiratory muscle weakness associated with hypophosphatemia (30). At present, there is little evidence of a link between phosphate depletion and clinically apparent muscle weakness.
Phosphate Replacement
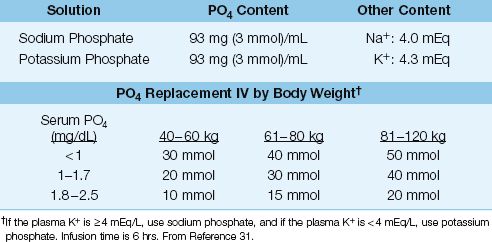
Intravenous phosphate replacement is recommended for all patients with severe hypophosphatemia (i.e., serum PO4 <1.0 mg/dL or 0.3 mmol/L), and for patients with hypophosphatemia of any degree who also have cardiac dysfunction, respiratory failure, muscle weakness, or impaired tissue oxygenation. The phosphate solutions and dosing regimen are shown in Table 38.5 (31).
Maintenance Therapy
The normal daily maintenance dose of phosphorus is 1,200 mg if given orally (32). The IV maintenance dose of PO4 is lower, at 800 mg/day, because only 70% of orally administered phosphate is absorbed from the GI tract.
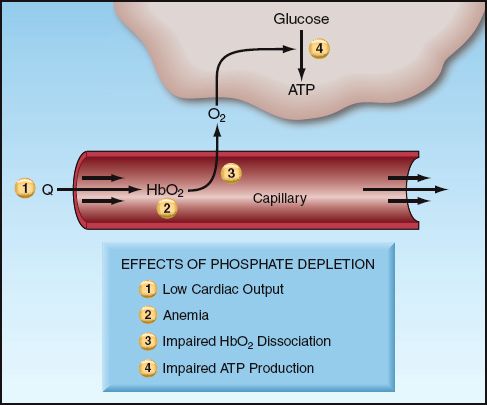
FIGURE 38.3 The effects of phosphate depletion that can impair energy utilization in cells. See text for explanation.
reTable 38.5 Phosphate Replacement Therapy
HYPERPHOSPHATEMIA
Most cases of hyperphosphatemia in the ICU are the result of impaired PO4 excretion from renal insufficiency, or PO4 release from disrupted cells (e.g., rhabdomyolysis or tumor lysis).
Clinical Manifestations
The clinical manifestations of hyperphosphatemia include: (a) the formation of insoluble calcium–phosphate complexes (with deposition into soft tissues), and (b) acute hypocalcemia (with tetany) (11). There is little information about the prevalence or significance of these manifestations in ICU patients.
Management
There are two approaches to hyperphosphatemia. The first is to promote PO4 binding in the upper GI tract, which can lower serum PO4 levels, even in the absence of any oral intake of phosphate (i.e., GI dialysis). Sucralfate or aluminum-containing antacids can be used for this purpose. In patients with significant hypocalcemia, calcium acetate tablets (PhosLo, Braintree Labs) can help raise the serum calcium while lowering the serum PO4. Each calcium acetate tablet (667 mg) contains 8.45 mEq elemental calcium. The recommended dose is 2 tablets three times a day (33,34).
The other approach to hyperphosphatemia is to enhance PO4 clearance with hemodialysis. This is reserved for patients with renal failure, and is rarely necessary.
A FINAL WORD
Despite the potential for harm, disorders of calcium and phosphorus are notable for the lack of apparent adverse consequences. It may be that abnormal plasma levels of calcium and phosphorus are markers of illness more than abnormalities that warrant correction. Otherwise, the following points about calcium and phosphorus deserve emphasis.
For calcium:
1. For patients with hypoalbuminemia, do not use any of the correction factors proposed for adjusting the plasma calcium concentration (because they are unreliable). You must measure the ionized calcium in these patients.
2. Magnesium depletion, which is common in ICU patients, should always be considered as a possible cause of hypocalcemia.
3. Because calcium infusions can be damaging, intravenous calcium should be reserved for cases of symptomatic hypocalcemia (which is uncommon, as mentioned).
For phosphorus:
1. Watch the plasma PO4 levels carefully when starting parenteral nutrition because of the risk for hypophosphatemia. This also applies to the use of continuous insulin infusions for tight glycemic control.
2. Watch for hypophosphatemia in patients receiving sucralfate for prophylaxis of stress ulcer bleeding.
REFERENCES
Calcium Reviews
1. Bushinsky DA, Monk RD. Electrolyte quintet: Calcium. Lancet 1998; 352:306–311.
2. Baker SB, Worthley LI. The essentials of calcium, magnesium and phosphate metabolism: part I. Physiology. Crit Care Resusc 2002; 4:301–306.
3. Weaver CM, Heaney RP. Calcium. In: Shils ME, et al., eds. Modern nutrition in health and disease. 10th ed. Philadelphia, PA: Lippincott, Williams & Wilkins, 2006; 194–210.
Plasma Calcium
4. Slomp J, van der Voort PH, Gerritsen RT, et al. Albumin-adjusted calcium is not suitable for diagnosis of hyper- and hypocalcemia in the critically ill. Crit Care Med 2003; 31:1389–1393.
5. Byrnes MC, Huynh K, Helmer SD, et al. A comparison of corrected serum calcium levels to ionized calcium levels among critically ill surgical patients. Am J Surg 2005; 189:310–314.
6. Forman DT, Lorenzo L. Ionized calcium: its significance and clinical usefulness. Ann Clin Lab Sci 1991; 21:297–304.
Ionized Hypocalcemia
7. Moritoki E, Kim I, Nichol A, et al. Ionized calcium concentration and outcome in critical illness. Crit Care Med 2011; 39:314–321.
8. Zaloga GP. Hypocalcemia in critically ill patients. Crit Care Med 1992; 20:251–262.
9. Burchard KW, Simms HH, Robinson A, et al. Hypocalcemia during sepsis. Relationship to resuscitation and hemodynamics. Arch Surg 1992; 127:265–272.
10. Steinberg W, Tenner S. Acute pancreatitis. N Engl J Med 1994; 330:1198–1210.
11. Baker SB, Worthley LI. The essentials of calcium, magnesium and phosphate metabolism: part II. Disorders. Crit Care Resusc 2002; 4:307–315.
12. Zaloga G. Divalent cations: calcium, magnesium, and phosphorus. In: Chernow B., ed. The pharmacologic approach to the critically ill patient. Baltimore: Williams & Wilkins, 1994.
13. Shapiro MJ, Mistry B. Calcium regulation and nonprotective properties of calcium in surgical ischemia. New Horiz 1996; 4:134–138.
14. Trump BF, Berezesky IK. Calcium-mediated cell injury and cell death. FASEB J 1995; 9:219–228.
Ionized Hypercalcemia
15. McCurdy MT, Shanholtz CB. Oncologic emergencies. Crit Care Med 2012; 40:2212–2222.
16. Stewart AF. Clinical practice. Hypercalcemia associated with cancer. N Engl J Med 2005; 352:373–379.
17. Body JJ. Hypercalcemia of malignancy. Semin Nephrol 2004; 24:48–54.
Hypophosphatemia
18. Geerse DA, Bindels AJ, Kuiper MA, et al. Treatment of hypophosphatemia in the intensive care unit: a review. Crit Care 2010; 14:R147. (An open access journal).
19. French C, Bellomo R. A rapid intravenous phosphate replacement protocol for critically ill patients. Critical Care Resusc 2004; 6:175–179.
20. Fiaccadori E, Coffrini E, Fracchia C, et al. Hypophosphatemia and phosphorus depletion in respiratory and peripheral muscles of patients with respiratory failure due to COPD. Chest 1994; 105:1392–1398.
21. Knochel JP. The pathophysiology and clinical characteristics of severe hypophosphatemia. Arch Intern Med 1977; 137:203–220.
22. Marinella MA. Refeeding syndrome and hypophosphatemia. J Intensive Care Med 2005; 20:155–159.
23. Paleologos M, Stone E, Braude S. Persistent, progressive hypophosphataemia after voluntary hyperventilation. Clin Sci 2000; 98:619–625.
24. Bodenhamer J, Bergstrom R, Brown D, et al. Frequently nebulized beta-agonists for asthma: effects on serum electrolytes. Ann Emerg Med 1992; 21:1337–1342.
25. Barak V, Schwartz A, Kalickman I, et al. Prevalence of hypophosphatemia in sepsis and infection: the role of cytokines. Am J Med 1998; 104:40–47.
26. Miller SJ, Simpson J. Medication-nutrient interactions: hypophosphatemia associated with sucralfate in the intensive care unit. Nutr Clin Pract 1991; 6:199–201.
27. King AL, Sica DA, Miller G, et al. Severe hypophosphatemia in a general hospital population. South Med J 1987; 80:831–835.
28. Davis SV, Olichwier KK, Chakko SC. Reversible depression of myocardial performance in hypophosphatemia. Am J Med Sci 1988; 295:183–187.
29. Agusti AG, Torres A, Estopa R, et al. Hypophosphatemia as a cause of failed weaning: the importance of metabolic factors. Crit Care Med 1984; 12:142–143.
30. Gravelyn TR, Brophy N, Siegert C, et al. Hypophosphatemia-associated respiratory muscle weakness in a general inpatient population. Am J Med 1988; 84:870–876.
31. Taylor BE, Huey WY, Buchman TG, et al. Treatment of hypophosphatemia using a protocol based on patient weight and serum phosphorus level in a surgical intensive care unit. J Am Coll Surg 2004; 198:198–204.
32. Knochel JP. Phosphorous. In: Shils ME, et al., eds. Modern nutrition in health and disease. 10th ed. Philadelphia, PA: Lippincott, Williams & Wilkins, 2006; 211–222.
Hyperphosphatemia
33. Kraft MD, Btaiche IF, Sacks GS, et al. Treatment of electrolyte disorders in adult patients in the intensive care unit. Am J Health Syst Pharm 2005; 62:1663–1682.
34. Lorenzo Sellares V, Torres Ramirez A. Management of hyperphosphataemia in dialysis patients: role of phosphate binders in the elderly. Drugs Aging 2004; 21:153–165.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


