FIGURE 23.1 Microscopic images of a normal lung and a lung in the advanced stages of ARDS, which shows a dense infiltration of leukocytes and proteinaceous material that fills and obliterates the normal architecture of the lungs.
Predisposing Conditions
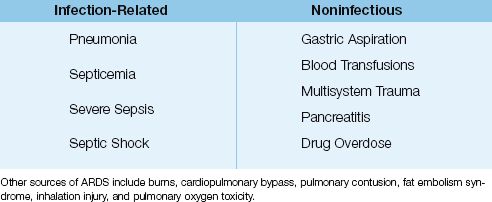
ARDS is not a primary disorder, but is a consequence of a variety of infectious and noninfectious conditions. The common conditions that predispose to ARDS are listed in Table 23.1. The most frequent offenders are pneumonia and the “sepsis syndromes” (i.e., septicemia, severe sepsis, and septic shock) (1,5). One feature shared by many of these conditions is the ability to trigger a systemic inflammatory response, which involves neutrophil activation, the principal inciting event in ARDS.
Table 23.1 Common Sources of ARDS
CLINICAL FEATURES
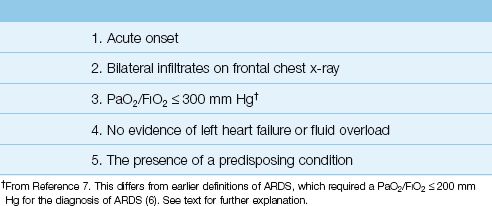
The clinical features of ARDS are listed in Table 23.2. The principal features include an acute onset, severe hypoxemia, and bilateral pulmonary infiltrates without evidence of left heart failure or volume overload. The earliest signs of ARDS are the sudden appearance of hypoxemia and signs of respiratory distress (e.g., dyspnea, tachypnea). The chest x-ray can be unrevealing in the first few hours after symptom onset, but bilateral pulmonary infiltrates begin to appear within 24 hours. Progressive hypoxemia requiring mechanical ventilation often occurs in the first 48 hours of the illness.
Table 23.2 Clinical Features of ARDS
Diagnostic Problems
Despite over 40 years of clinical experience with ARDS, there is still some uncertainty about the defining features of this condition. In 1994, a consensus conference of experts published a set of diagnostic criteria for ARDS and a clinical entity known as acute lung injury (ALI) (6). These criteria included: (a) PaO2/FIO2 ″200 mm Hg for ARDS, (b) PaO2/FIO2 ″300 mm Hg for ALI, and (c) a pulmonary artery wedge pressure (PAWP) ″18 mm Hg (to rule out left heart failure). In 2012, a European task force published a set of revised criteria for the diagnosis of ARDS (7) that included the following changes: (a) ALI was eliminated as a clinical entity, and the PaO2/FIO2 for ARDS was set at ″300 mm Hg, (b) a requirement was added that the PaO2/FIO2 determination should be conducted at a positive end-expiratory pressure (PEEP) of 5 cm H2O, and (c) the wedge pressure measurement was eliminated (because of the diminished use of pulmonary artery catheters). These revised criteria are known as the Berlin Criteria, and they are combined with the original diagnostic criteria for ARDS in Table 23.2. Not included is the requirement for a standard level of PEEP during the PaO2/FIO2 determination because PEEP requires mechanical ventilation, and the diagnosis of ARDS can occur during spontaneous breathing.
Lack of Specificity
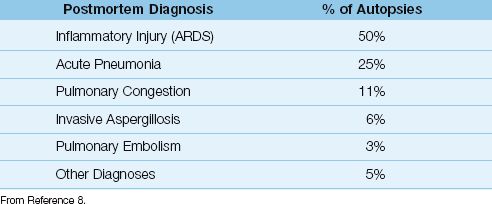
Many of the diagnostic criteria for ARDS are non-specific, and are shared by other common causes of acute respiratory failure. This creates a tendency for misdiagnosis, which is demonstrated in Table 23.3 (8). The information in this table is from an autopsy study of patients who died with a premortem diagnosis of ARDS, and the postmortem diagnoses are listed in the table along with the prevalence of each diagnosis. Only half of the patients with the premortem diagnosis of ARDS had evidence of ARDS on postmortem examination, and the conditions most commonly mistaken for ARDS were pneumonia and hydrostatic pulmonary edema. The likelihood of identifying ARDS was 50% in this study, which is no better than the likelihood of heads or tails with a coin flip!
Table 23.3 Postmortem Diagnosis in Patients with a Premortem Diagnosis of ARDS
Radiographic Appearance
One source of error in the diagnosis of ARDS is the appearance of the chest x-ray. The classic radiographic appearance of ARDS is shown in Figure 23.2. The infiltrate has a finely granular or ground-glass appearance, and is evenly distributed in all lung fields, with no evidence of a pleural effusion. Unfortunately, these characteristic features are not always present. This is demonstrated by the chest x-ray in Figure 23.3. In this case, the infiltration shows a hilar prominence and is confined to the lower lung fields, with obliteration of the left hemidiaphragm suggesting a possible pleural effusion. These features could be mistaken for cardiogenic pulmonary edema. Because of such variability in the radiographic ap-pearance of ARDS, it is not possible to identify ARDS reliably using the chest x-ray alone (9).
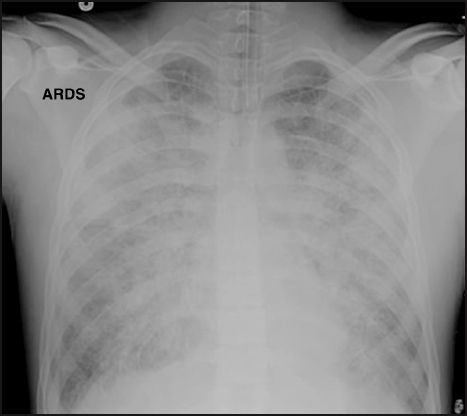
FIGURE 23.2 Portable chest x-ray showing the classic radiographic appearance of ARDS. The infiltrate has a finely granular or “ground glass” appearance, and is evenly distributed throughout both lungs, with a relative sparing of the lung bases. There is no evidence of a pleural effusion.
Pitfalls of the Wedge Pressure
When the chest-x-ray shows overlapping features of ARDS and cardiogenic pulmonary edema, like the one in Figure 23.3, the pulmonary artery wedge pressure (PAWP) has been used to distinguish between these two conditions (i.e., a PAWP ″18 mm Hg is considered evidence of ARDS) (6). This is problematic because the wedge pressure is not a measure of capillary hydrostatic pressure, as explained in Chapter 8 (see pages 140-141). The PAWP is measured in the absence of blood flow, when the static column of blood between the catheter tip and left atrium results in an equalization of pressures between the wedge pressure and left atrial pressure. However, when flow resumes, the pressure in the pulmonary capillaries must be higher than the left atrial pressure to provide a pressure gradient for flow in the pulmonary veins. Therefore, the left atrial (wedge) pressure is lower than the capillary hydrostatic pressure, and this will lead to an overdiagnosis of ARDS.
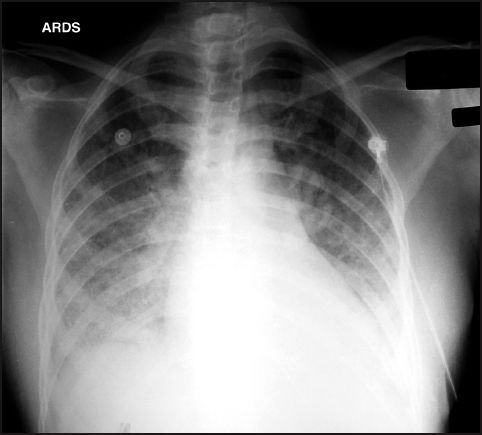
FIGURE 23.3 Portable chest x-ray of ARDS in a patient with urosepsis and Gram-negative septicemia. The infiltrates appear to emanate from the hilar areas and are confined to the lower lung fields. There is also obliteration of the left hemidiaphragm suggesting the presence of a pleural effusion. These radiographic features could be mistaken for hydrostatic pulmonary edema.
Bronchoalveolar Lavage
Although rarely used, bronchoalveolar lavage is a reliable method for distinguishing ARDS from cardiogenic pulmonary edema (10). This procedure is performed at the bedside using a flexible fiberoptic bronchoscope that is advanced into one of the involved lung segments. Once in place, the lung segment is lavaged with isotonic saline, and the lavage fluid is analyzed for the presence of neutrophils and protein.
1. In normal subjects, neutrophils make up less than 5% of the cells recovered in lung lavage fluid, whereas in patients with ARDS, as many as 80% of the recovered cells are neutrophils (10). A low neutrophil count in lung lavage fluid can be used to exclude the diagnosis of ARDS, while a high neutrophil count is evidence of ARDS.
2. Because inflammatory exudates are rich in proteinaceous material, lung lavage fluid that is rich in protein is used as evidence of ARDS. When the protein concentration in lung lavage fluid is expressed as a fraction of the protein concentration in plasma, the following criteria can be applied (11):
Hydrostatic Edema:Lavage fluid [protein] / plasma [protein] <0.5
ARDS:Lavage fluid [protein] / plasma [protein] >0.7
VENTILATOR MANAGEMENT IN ARDS
One of the most important discoveries in critical care medicine in the last quarter-century is the role of mechanical ventilation as a source of lung injury, particularly in patients with ARDS. This has led to a management strategy known as lung protective ventilation (12), which is described here.
Conventional Mechanical Ventilation
Since the introduction of positive-pressure mechanical ventilation, the use of large inflation volumes (tidal volumes) has been a standard practice to reduce the tendency for atelectasis during mechanical ventilation. The conventional tidal volumes are 12 to 15 mL/kg (13), which are twice the size of tidal volumes achieved during quiet breathing (6 to 7 mL/kg). In patients with ARDS, these large inflation volumes are delivered into lungs that have only a fraction of the normal functional lung volume, as described next.
Functional Volume in ARDS
Although portable chest x-rays show an apparent homogeneous pattern of lung infiltration in ARDS, CT images reveal that the lung infiltration in ARDS is confined to dependent lung regions (13). This is shown in the CT images in Figure 23.4. Note the dense consolidation in the posterior lung regions (which are the dependent lung regions in the supine position), and the normal or uninvolved lung is restricted to the anterior half of the thorax. The uninvolved areas represent the functional portion of the lungs, and the portion that receives the inflation volumes. Therefore, the high inflation volumes used during conventional mechanical ventilation are being delivered to a markedly reduced volume of available lung, and this results in overdistension and rupture of the distal airspaces (15).
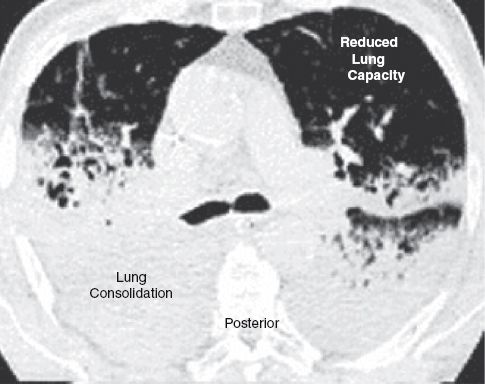
FIGURE 23.4 Computed tomographic image of lung slices in the region of the hilum from a patient with ARDS. The lung consolidation is confined to the posterior lung regions, which are the dependent regions in the supine position. The uninvolved lung in the anterior one-third of the thorax represents the functional portion of the lung. CT image is from Reference 14, and is digitally retouched.
Ventilator-Induced Lung Injury
Excessive inflation of the distal airspaces produce stress fractures in the alveolar capillary interface, and this leads to infiltration of the lung parenchyma and distal airspaces with an inflammatory exudate. This condition is known as ventilator-induced lung injury (VILI), and is strikingly similar to ARDS (15). The lung injury is volume-related rather than pressure-related (16), and is called volutrauma. (Pressure-related lung injury is called barotrauma, and is associated with the escape of air from the lungs.)
BIOTRAUMA: During conventional, high-volume mechanical ventilation, proinflammatory cytokines can appear in the lungs and systemic circulation, even though there is no structural damage in the lungs (17). This proinflammatory condition is known as biotrauma, and it can lead to neutrophil activation and inflammatory infiltration in the lungs (16). The systemic inflammatory response associated with biotrauma can promote inflammatory injury in other organs, which means that mechanical ventilation can be as source of inflammatory-mediated multiorgan failure (18)!
ATELECTRAUMA: The decrease in lung distensibility in ARDS can result in the collapse of small airways at the end of expiration. When this occurs, mechanical ventilation can be associated with cyclic opening and closing of small airways, and this process can be a source of lung injury (19). This type of lung injury is called atelectrauma (16), and it may be the result high-velocity shear forces created by the opening of collapsed airways, which can damage the airway epithelium.
Lung Protective Ventilation
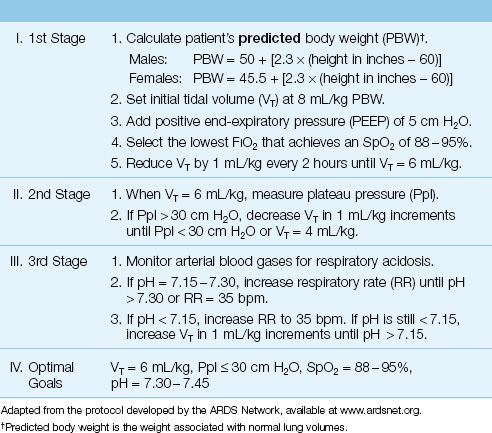
Lung protective ventilation employs low tidal volumes (6 mL/kg) to limit the risk of volutrauma and biotrauma, and uses positive end-expiratory pressure (PEEP) to limit the risk of atelectrauma. A protocol for lung protective ventilation that has a proven survival benefit in ARDS (20) is shown in Table 23.4. This protocol was developed by the ARDS Clinical Network (a network created by governmental health agencies to evaluate potential therapies for ARDS), and is available at www.ardsnet.org. The tidal volume in this protocol is 6 mL/kg, based on predicted body weight, which is the body weight associated with normal lung volumes. Note that one of the stated goals is an end-inspiratory “plateau” pressure (Ppl) ″30 cm H2O. This pressure is described in detail in Chapter 25.
Table 23.4 Protocol for Lung Protective Ventilation in ARDS
Positive End-Expiratory Pressure
(For a detailed description of this pressure, see Chapter 26.) Lung protective ventilation employs a positive end-expiratory pressure (PEEP) of at least 5 cm H2O to prevent the collapse of small airways at the end of expiration. The goal is to prevent the cyclic opening and closing of small airways and reduce the risk of atelectrauma. Higher PEEP levels (e.g., 15 cm H2O) have been associated with a shorter duration of mechanical ventilation and a borderline increase in survival in ARDS, but only when the PaO2/FIO2 ratio is ″200 mm Hg (21). However, PEEP levels above 10 cm H2O are not typically used unless there is a problem maintaining arterial oxygenation (see next).
In situations where a toxic level of inhaled oxygen (FIO2 >50%) is needed to maintain the target SpO2 of 88–95%, PEEP levels above 5 cm H2O can be used to improve arterial oxygenation and reduce the FIO2 to safer levels. However, it is important to emphasize that increases in PEEP can reduce the cardiac output, and if the goal of increasing PEEP is to maintain the same SpO2 at a lower FIO2, the reduced cardiac output will reduce the systemic O2 delivery.
Permissive Hypercapnia
One of the consequences of low tidal volume ventilation is a decrease in CO2 elimination in the lungs, which can result in hypercapnia and respiratory acidosis. Because of the benefits of low volume ventilation, hypercapnia is allowed to persist as long as there is no evidence of harm. This practice is known as permissive hypercapnia (22). The limits of tolerance to hypercapnia and respiratory acidosis are unclear, but data from clinical trials of permissive hypercapnia show that arterial PCO2 levels of 60–70 mm Hg and arterial pH levels of 7.2–7.25 are safe for most patients (23). The target pH is 7.30–7.45 in the protocol for lung protective ventilation in Table 23.4.
Impact on Survival
Lung protective ventilation is one of the few measures that has been shown to improve survival in ARDS. The largest and most successful trial of lung protective ventilation was conducted by the ARDS Network (20), and enrolled over 800 ventilator-dependent patients with ARDS who were randomly assigned to receive tidal volumes of 6 mL/kg or 12 mL/kg (using predicted body weight). Ventilation with the lower tidal volume (6 mL/kg), and an end-inspiratory plateau pressure (Ppl) ″30 cm H2O, was associated with a shorter duration of mechanical ventilation and a 9% absolute reduction in mortality rate (40% to 31%, P=0.007).
A total of 5 clinical trials have compared tidal volumes of 6 mL/kg and 12 mL/kg during mechanical ventilation in patients with ARDS. In two of the trials, low tidal volumes were associated with fewer deaths, while in three of the trials, there was no survival benefit associated with low tidal volumes (24). Despite the lack of a consistent survival benefit, lung protective ventilation using tidal volumes of 6 mL/kg has become a standard practice in patients with ARDS. A recent multicenter survey of lung protective ventilation in ARDS showed an in-hospital mortality rate of 48% (5), which is no better than mortality rates reported before the introduction of lung protective ventilation. An observation that is relevant in this context is that mortality rates for ARDS tend to be lower in controlled clinical trials than in clinical practice surveys (25).
(Note: A possible explanation for the lack of a consistent survival benefit with low tidal volumes is presented in Chapter 25.)
Summary
There is convincing evidence that mechanical ventilation can damage the lungs in ARDS as a result of overdistension of functional alveoli (volutrauma) and collapse of small airways (atelectrauma). Lung protective ventilation is designed to mitigate the mechanical forces that create ventilator-induced lung injury, and it has been adopted as a standard method of mechanical ventilation in ARDS.
NONVENTILATORY MANAGEMENT
The treatment of ARDS begins by treating the inciting condition (e.g., septicemia), if possible. Therapies directed at the ARDS have been marked by failure more than success. The failed therapies in ARDS in-clude surfactant (in adults), inhaled nitric oxide, pentoxyphylline, ibuprofen, prostaglandin E1, and antifungal agents (to inhibit thromboxane) (26). Clinical benefits have been reported with fluid management that avoids fluid accumulation in the lungs, and with high-dose corticosteroids in severe or unresolving ARDS. The management described in this section is limited to the measures that have documented benefits.
Fluid Management
The lung consolidation in ARDS is an inflammatory exudate, and should not be influenced by fluid balance (for the same reason that diuresis will not clear an infiltrate caused by pneumonia). However, avoiding a positive fluid balance will prevent unwanted fluid accumulation in the lungs, which could aggravate the respiratory insufficiency in ARDS. Clinical studies have shown that avoiding a positive fluid balance in patients with ARDS can reduce the time on mechanical ventilation (27), and can even reduce mortality (28).
However, it is also important to avoid fluid deficits and maintain intra-vascular volume because the positive intrathoracic pressures during mechanical ventilation will magnify the tendency for the cardiac output to decrease in response to deficits in intravascular volume.
Corticosteroid Therapy
There is a long history of clinical trials evaluating steroid therapy in ARDS, and the aggregated results of these studies show no consistent survival benefit associated with steroid therapy (29). However, there is evidence of other benefits provided by steroid therapy in ARDS, and these include a reduction in markers of inflammation (both pulmonary and systemic inflammation), improved gas exchange, shorter duration of mechanical ventilation, and shorter length of stay in the ICU (29). Steroid therapy is currently recommended only in cases of early severe ARDS and unresolving ARDS (28).
Early Severe ARDS
In early severe ARDS, defined as a PaO2/FIO2 <200 mm Hg with PEEP of 10 cm H2O, the following steroid regimen in recommended (29):
Methylprednisolone: Start with an IV loading dose of 1 mg/kg (ideal body weight) over 30 minutes, then infuse at 1 mg/kg/day for 14 days, then gradually taper the dose over the next 14 days and discontinue therapy. Five days after the patient is able to ingest oral medications, the dose can be given orally (as prednisone or prednisolone) as a single daily dose.
Unresolving ARDS
ARDS has a fibrinoproliferative phase that begins 7–14 days after the onset of illness (30), and eventually results in irreversible pulmonary fibrosis. High-dose steroid therapy started in the developing phase of fibrinoproliferation can help to halt the progression to pulmonary fibrosis. In cases where ARDS does not begin to resolve after 7 days, high-dose steroid therapy is recommended, but should begin no later than 14 days after the onset of illness. The following steroid regimen is recommended (29):
Methylprednisolone: Start with an IV loading dose of 2 mg/kg (ideal body weight) over 30 minutes, then infuse at 2 mg/kg/day for 14 days, and 1 mg/kg/day for the next 7 days. After this, gradually taper the dose and discontinue therapy at 2 weeks after extubation. Five days after the patient is able to ingest oral medications, the dose can be given orally (as prednisone or prednisolone) as a single daily dose.
The risks of high-dose steroid therapy include worsening glycemic control and prolonged neuromuscular weakness when combined with neuromuscular blocking agents. There is no evidence of an increased risk of nosocomial infections with the steroid regimens described here (28).
Therapeutic Misdirection?
Although the therapeutic focus for ARDS has been the lungs, the principal cause of death in ARDS is multiorgan failure, not respiratory failure (5,31). As many as 70% of deaths in ARDS are the result of multiorgan failure (31), and the mortality rate is directly related to the number of organs that fail. The relationship between mortality rate and extrapulmonary organ failure is shown in Figure 23.5. This relationship indicates that mortality is a function of a progressive systemic condition, which most likely represents progressive systemic inflammation. The similarities between Figure 23.5 and Figure 14.2 (see page 267) supports the notion of a relationship between progressive systemic inflammation and mortality in ARDS. If this is the case, then limiting the therapeutic focus to the lungs in ARDS is a prescription for failure.
REFRACTORY HYPOXEMIA
A minority (10 to 15%) of patients with ARDS develop severe hypoxemia that is refractory to oxygen therapy and mechanical ventilation (32). This condition is an immediate threat to life, and the following “rescue therapies” can produce an immediate improvement in arterial oxygenation. Unfortunately, these rescue measures often provide little or no survival benefit.
High Frequency Oscillatory Ventilation
High frequency oscillatory ventilation (HFOV) delivers small tidal volumes (1–2 mL/kg) using rapid pressure oscillations (300 cycles/min). The small tidal volumes limit the risk of volutrauma, and the rapid pressure oscillations create a mean airway pressure that prevents small airway collapse and limits the risk of atelectrauma. When used in patients with severe ARDS, HFOV can improve arterial oxygenation, but there is no documented survival benefit (33). HFOV requires a specialized ventilator (Sensormedics 3100B, Viasys Healthcare, Yorba Linda, CA), and may not be available in all hospitals. This mode of ventilation is de-scribed in more detail in Chapter 27.
Inhaled Nitric Oxide
Inhaled nitric oxide (5–10 ppm) is a selective pulmonary vasodilator that can improve arterial oxygenation in ARDS by increasing flow to areas of high dead space ventilation (34). However, the increase in arterial oxygenation is temporary (1–4 days), and there is no associated survival benefit (35). Adverse effects of inhaled nitric oxide include methemoglobinemia (usually mild) and renal dysfunction (35). An added risk is the potential for nitric oxide to form peroxinitrite, a potent toxin capable of oxidant cell injury.
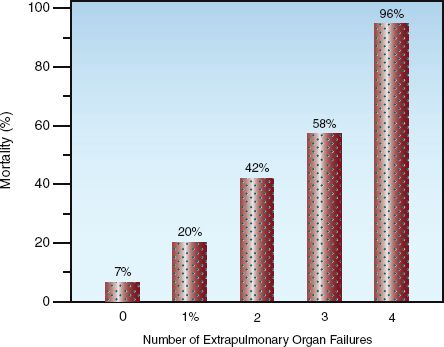
FIGURE 23.5 The relationship between extrapulmonary organ failure and mortality in ARDS. Data from the electronic supplementary material for Reference 5.
Prone Position
Switching from the supine to prone position can improve pulmonary gas exchange by diverting blood away from poorly aerated lung regions in the posterior thorax and increasing blood flow in aerated lung regions in the anterior thorax (see Figure 23.4). Prone positioning has had little impact on mortality in ARDS, but a recent study combining lung protective ventilation with prone positioning showed a lower than expected mortality rate in patients with severe ARDS (PaO2/FIO2 <100 mm Hg) (36). Prone positioning is labor intensive and creates problems with nursing care (e.g., airway care and skin care), but it may be the only measure available for refractory hypoxemia in hospitals with limited resources.
ECMO
Extracorporeal membrane oxygenation (ECMO) has had variable success in patients with refractory hypoxemia, and is a consideration only in medical centers with established ECMO programs, and only when other rescue therapies have failed (37).
A FINAL WORD
The Evils of Inflammation
One of the important messages in this book is the prominent role of inflammation as a source of organ damage and multiorgan failure in critically ill patients, and the lack of an effective treatment for this destructive force. This message appears in Chapter 14, which describes inflammatory shock and multiorgan failure, and it appears again in this chapter. Although a survival benefit has been documented with lung protective ventilation in ARDS, this is not a treatment for ARDS, but rather is a lessening of the damage produced by mechanical ventilation. Until a remedy is available for the destructive effects of inflammation, conditions like ARDS will continue to be a major source of morbidity and mortality in the ICU.
REFERENCES
Review
1. Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest 2012; 122:2731–2740.
Pathogenesis
2. Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. Lancet 1967; 2:319–323.
3. Abraham E. Neutrophils and acute lung injury. Crit Care Med 2003; 31(Suppl): S195–S199.
4. Idell S. Coagulation, fibrinolysis, and fibrin deposition in acute lung injury. Crit Care Med 2003; 31(Suppl):S213–S220.
5. Villar J, Blanco J, Anon JM, et al. The ALIEN study: incidence and outcome of acute respiratory distress syndrome in the era of lung protective ventilation. Intensive Care Med 2011; 37:1932–1941.
Clinical Features
6. Bernard GR, Artigas A, Brigham KL, et al. The American–European Consen-sus Conference on ARDS: definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am Rev Respir Crit Care Med 1994; 149:818–824.
7. The ARDS Definition Task Force. Acute respiratory distress syndrome. The Berlin definition. JAMA 2012; 307:2526–2533.
8. de Hemptinne Q, Remmelink M, Brimioulle S, et al. ARDS: A clinicopathological confrontation. Chest 2009; 135:944–949.
9. Rubenfeld GD, Caldwell E, Granton J, et al. Interobserver variability in applying a radiographic definition for ARDS. Chest 1999; 116:1347–1353.
10. Idell S, Cohen AB. Bronchoalveolar lavage in patients with the adult respiratory distress syndrome. Clin Chest Med 1985; 6:459–471.
11. Sprung CL, Long WM, Marcial EH, et al. Distribution of proteins in pulmonary edema. The value of fractional concentrations. Am Rev Respir Dis 1987; 136:957–963.
Ventilator Management in ARDS
12. Brower RG, Rubenfeld GD. Lung-protective ventilation strategies in acute lung injury. Crit Care Med 2003; 31(Suppl):S312–S316.
13. Pontoppidan H, Geffen B, Lowenstein E. Acute respiratory failure in the adult. N Engl J Med 1972; 287:799–806.
14. Rouby J-J, Puybasset L, Nieszkowska A, Lu Q. Acute respiratory distress syndrome: Lessons from computed tomography of the whole lung. Crit Care Med 2003; 31(Suppl):S285–S295.
15. Dreyfuss D, Saumon G. Ventilator-induced lung injury. Am J Respir Crit Care Med 1998; 157:294–323.
16. Gattinoni L, Protti A, Caironi P, Carlesso E. Ventilator-induced lung injury: the an-atomical and physiological framework. Crit Care Med 2010; 38(Suppl):S539–S548.
17. Ranieri VM, Suter PM Tortorella C, et al. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: A randomized controlled trial. JAMA 1999; 282:54–61.
18. Ranieri VM, Giunta F, Suter P, Slutsky AS. Mechanical ventilation as a mediator of multisystem organ failure in acute respiratory distress syndrome. JAMA 2000; 284:43–44.
19. Muscedere JG, Mullen JBM, Gan K, et al. Tidal ventilation at low airway pressures can augment lung injury. Am J Respir Crit Care Med 1994; 149:1327–1334.
20. The Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. New Engl J Med 2000; 342:1301–1308.
21. Briel M, Meade M, Mercat A, et al. Higher vs. lower positive end-expiratory pressure in patients with acute lung injury and acute respiratory distress syndrome. JAMA 2010; 303:865–873.
22. Bidani A, Tzouanakis AE, Cardenas VJ, Zwischenberger JB. Permissive hypercapnia in acute respiratory failure. JAMA 1994; 272:957–962.
23. Hickling KG, Walsh J, Henderson S, et al. Low mortality rate in adult respiratory distress syndrome using low-volume, pressure-limited ventilation with permissive hypercapnia: A prospective study. Crit Care Med 1994; 22:1568–1578.
24. Fan E, Needham DM, Stewart TE. Ventilator management of acute lung injury and acute respiratory distress syndrome. JAMA 2005; 294:2889–2896.
25. Phua J, Badia JR, Adhikari NKJ, et al. Has mortality from acute respiratory distress syndrome decreased over time? Am J Respir Crit Care Med 2009; 179:220–227.
Nonventilatory Management
26. Calfee CS, Matthay MA. Nonventilatory treatment for acute lung injury and ARDS. Chest 2007; 131:913–920.
27. The Acute Respiratory Distress Syndrome Network. Comparison of two fluid management strategies in acute lung injury. N Engl J Med 2006; 354:2564–2575.
28. Murphy CV, Schramm GE, Doherty JA, et al. The importance of fluid management in acute lung injury secondary to septic shock. Chest 2009; 136:102–109.
29. Marik PE, Meduri GU, Rocco PRM, Annane D. Glucocorticoid treatment in acute lung injury and acute respiratory distress syndrome. Crit Care Clin 2011; 27:589–607.
30. Meduri GU, Chinn A. Fibrinoproliferation in late adult respiratory distress syndrome. Chest 1994; 105(Suppl):127S–129S.
31. Estenssoro E, Dubin A, Laffaire E, et al. Incidence, clinical course, and outcome in 217 patients with acute respiratory distress syndrome. Crit Care Med 2002; 30:2450–2456.
Refractory Hypoxemia
32. Pipeling MR, Fan E. Therapies for refractory hypoxemia in acute respiratory distress syndrome. JAMA 2010; 304:2521–2527.
33. Stawicki SP, Goyal M, Sarani B. High-frequency oscillatory ventilation (HFOV) and airway pressure release ventilation (APRV): A practical guide. J Intensive Care Med 2009; 24:215–229.
34. Griffiths MJ, Evans TW. Inhaled nitric oxide therapy in adults. N Engl J Med 2005; 353:2683–2695.
35. Adhikari NK, Burns KE, Friedrich JO, et al. Effect of nitric oxide on oxygenation and mortality in acute lung injury: systematic review and meta-analysis. British Med J 2007; 334(7597):779–787.
36. Charron C, Bouferrache K, Caille V, et al. Routine prone positioning in patients with severe ARDS: feasibility and impact on prognosis. Intensive Care Med 2011; 37:785–790.
37. Raoof S, Goulet K, Esan A, et al. Severe hypoxemic respiratory failure: Part 2 – Nonventilatory strategies. Chest 2010; 137:1437–1448.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


