FIGURE 54.1 Prescription drug use in the United States in 2007–2010, by age group. Data from Health United States, 2012. DHHS Publication No. 2013-1232, May, 2013.
Toxic Mechanism
The toxicity of acetaminophen is related to its metabolism in the liver, and the metabolic pathways are shown Figure 54.2. The bulk of acetaminophen metabolism involves the formation of nontoxic sulfated conjugates, which are excreted in the urine. About 10% of the metabolism involves the oxidation of acetaminophen to form a toxic metabolite that is capable of producing oxidant cell injury. When the daily dose of acetaminophen is not excessive, the toxic metabolite is removed by conjugation with glutathione, an intracellular antioxidant. When the daily dose of acetaminophen exceeds 4 grams, the sulfated conjugation pathways become saturated, which diverts acetaminophen metabolism to the pathway with the toxic metabolite. The increased traffic in this pathway eventually depletes glutathione reserves, and when glutathione stores fall to 30% of normal, the toxic acetaminophen metabolite can accumulate and promote hepatocellular damage (3).
Toxic Dose
The toxic dose of acetaminophen can vary widely in individual patients, but is somewhere between 7.5 and 15 grams in most adults (6,7). However, several conditions can increase the susceptibility to acetaminophen hepatotoxicity, such as malnutrition, chronic illness, and chronic ethanol ingestion, and the toxic dose of acetaminophen may be lower in these patients. A general rule adopted by poison control centers is to recommend evaluation for acute ingestions of 10 grams of acetaminophen (20 extra-strength Tylenol tablets) (3). For patients who may have an increased susceptibility to acetaminophen hepatotoxicity, an acute ingestion of 4 grams of acetaminophen warrants an evaluation (3). The maximum recommended daily dose of acetaminophen is 3.25 grams (5).
Clinical Presentation
In the first 24 hours following a toxic ingestion, symptoms are either absent or nonspecific (e.g., nausea, vomiting, malaise), and liver enzymes do not begin to rise until 24–36 hours post-ingestion (6). Elevated aspartate aminotranferase (AST) is the most sensitive marker of acetaminophen toxicity; the rise in AST precedes the hepatic dysfunction, and peak levels are reached at 72–96 hours. Evidence of hepatic injury becomes apparent 24–48 hours after ingestion, with steadily rising liver enzymes, and the appearance of jaundice and a coagulopathy. Peak hepatic injury occurs at 3–5 days after the toxic ingestion, and hepatic encephalopathy may be evident at this time, along with acute, oliguric renal failure (mechanism unclear), and lactic acidosis (from reduced hepatic clearance) (3). Death from hepatic injury usually occurs 3–5 days post-ingestion. Patients who survive often recover completely, although recovery can be prolonged.
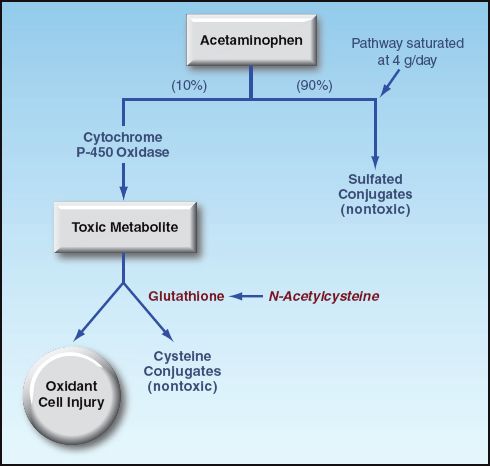
FIGURE 54.2 The hepatic metabolism of acetaminophen. See text for explanation.
Risk Assessment
In many cases of acetaminophen overdose, the initial encounter with the patient occurs within 24 hours after drug ingestion, when there are no manifestations of hepatic injury. The principal task at this time is to determine the risk of hepatocellular injury, and this involves two considerations: the time elapsed from the toxic ingestion, and the plasma level of acetaminophen. The ingested dose is not used to determine hepatotoxic risk because it is not possible to determine if the patient’s recollection of the ingested dose is accurate. In addition, the toxic dose of acetaminophen can vary in individual patients.
Plasma Drug Levels
Plasma acetaminophen levels obtained from 4–24 hours after drug ingestion can be used to predict the risk of hepatotoxicity using the nomogram in Figure 54.3 (7). If the plasma level is in the high-risk region of the nomogram, the risk of developing hepatotoxicity is 60% or higher, and antidote therapy is warranted. The risk of hepatotoxicity is only 1–3% in the low-risk region of the nomogram, and this does not warrant antidote therapy (7). This nomogram is useful only when the time of drug ingestion can be identified, and when the plasma drug level can be measured between 4 and 24 hours post-ingestion.
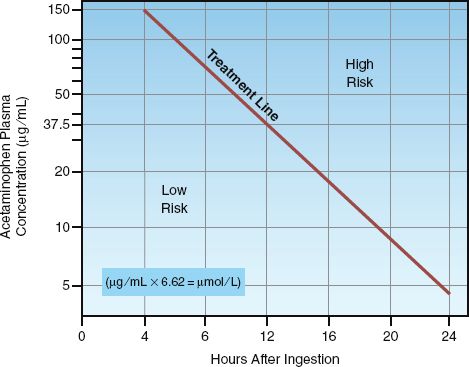
FIGURE 54.3 Nomogram for predicting the risk of hepatotoxicity according to the plasma acetaminophen level between 4 and 24 hours after ingestion. A plasma level that falls on, or above, the treatment line is an indication to begin antidotal therapy with N-acetylcysteine. Nomogram redrawn from Reference 7.
N-Acetylcysteine
Glutathione does not readily cross cell membranes, so exogenous glutathione is not a viable treatment option for acetaminophen hepatotoxicity. N-acetylcysteine (NAC) is a glutathione analogue that readily crosses cross cell membranes, and can serve as an intracellular glutathione surrogate (see Figure 54.2) (8). The cysteine residue in NAC contains a sulfhydryl group (SH), which is capable of reducing (and inactivating) the toxic acetaminophen metabolite.
Timing
NAC is most effective when therapy is started within 8 hours after ingestion; in this situation, NAC reduces the risk of hepatotoxicity to <5% (3). NAC is less protective after 10 hours have elapsed, but some protection is provided for 24 hours after drug ingestion (6,9). NAC can also have beneficial effects after the onset of acetaminophen hepatotoxicity (3,10). Therefore, evidence of hepatotoxicity after 24 hours post-ingestion is an indication to begin NAC therapy (3).
Treatment Regimens
NAC can be given intravenously or orally using the dosing regimens shown in Table 54.1. Despite significant differences in total NAC dose and duration of therapy, the two dosing regimens are considered equally effective (13,14). The intravenous route is preferred because it is the most reliable mode of drug delivery, and is not as disagreeable as oral ingestion of NAC (see “Adverse Reactions”).
EXTENDED TREATMENT: The standard duration of treatment is 21 hours for the IV regimen, and 72 hours for the oral regimen. However, because NAC can hasten the resolution of acetaminophen-induced liver injury (3), treatment with NAC should be continued beyond the normal course of therapy if there is evidence of continued liver injury (3). For the IV regimen, therapy is continued using the recommendations for the last 16 hours of the regimen. NAC can be discontinued when ALT levels have peaked and are improving, and when the INR is <1.3 (3).
Adverse Reactions
Intravenous NAC can cause anaphylactoid reactions, and fatal reactions have been reported in asthmatics (15). Oral NAC has a very disagreeable taste (because of the sulfur content), and often causes nausea and vomiting. The oral NAC regimen produces diarrhea in about 50% of patients, but this usually resolves with continued therapy (16).
Activated Charcoal
Acetaminophen is rapidly absorbed from the GI tract, and activated charcoal (1 g/kg body weight) is recommended only if given within the first 4 hours after drug ingestion (17). However, following massive drug ingestions, charcoal can provide a benefit when given as late as 16 hours after drug ingestion (3). Activated charcoal does not curb the efficacy of oral NAC (3).
Table 54.1 Treatment of Acetaminophen Overdose with N-Acetylcysteine (NAC)

BENZODIAZEPINES
Benzodiazepines are second only to opiates as the drugs most frequently involved in medication-related deaths (2). However, benzodiazepines are rarely fatal when ingested alone (18), and other respiratory depressant drugs (e.g., opiates) are almost always involved in benzodiazepine-related fatalities (2).
Clinical Features
Because overdoses involving benzodiazepines also involve other drugs, the clinical presentation can vary (according to the drugs ingested). Pure benzodiazepine overdoses produce deep sedation, but rarely result in coma (18). Respiratory depression (2–12% of cases), bradycardia (1–2% of cases) and hypotension (5–7% of cases) are also uncommon (18). Benzodiazepine intoxication can also produce an agitated confusional state (with hallucinations) that could be mistaken for alcohol withdrawal (18).
Involvement of benzodiazepines in an apparent overdose can be difficult to establish because there are no serum assays for benzodiazepines, and qualitative tests for benzodiazepines in urine are unreliable because of a limited spectrum of detection (19). Benzodiazepine involvement is usually based on the clinical history.
Management
The management of benzodiazepine overdose involves general supportive care, although an antidote is available.
Flumazenil
Flumazenil is a benzodiazepine antagonist that binds to benzodiazepine receptors, but does not exert any agonist actions (20). It is effective in reversing benzodiazepine-induced sedation, but is inconsistent in reversing benzodiazepine-induced respiratory depression (21).
DOSING REGIMEN: Flumazenil is given as an intravenous bolus. The initial dose is 0.2 mg, and this can be repeated at 1–6 minute intervals, if necessary, to a cumulative dose of 1 mg. The response is rapid, with onset in 1–2 minutes, and peak effect at 6–10 minutes (22). The effect lasts about one hour. Since flumazenil has a shorter duration of action than the benzodiazepines, sedation can return after 30–60 minutes. To reduce the risk of re-sedation, the initial bolus dose of flumazenil is often followed by a continuous infusion at 0.3–0.4 mg/hr (23).
ADVERSE REACTIONS: Flumazenil is safe to use in most patients. It can precipitate a benzodiazepine withdrawal syndrome in patients with a longstanding history of benzodiazepine use, but this is uncommon (24). Flumazenil can also precipitate seizures (a) in patients receiving benzodiazepines for seizure control, and (b) in mixed overdoses involving tricyclic antidepressants (25).
CLINICAL USE: Despite its effectiveness in reversing benzodiazepine-induced sedation, flumazenil is not a popular antidote. This is partly due to concerns about the risk of benzodiazepine withdrawal or seizures, and is partly due to the fact that benzodiazepine overdoses are rarely life-threatening.
ß-RECEPTOR ANTAGONISTS
Intentional β-blocker overdoses are uncommon, but can be life-threatening. An effective antidote is available, if needed.
Toxic Manifestations
The typical manifestations of β-blocker overdose are bradycardia and hypotension (26). The bradycardia is usually sinus in origin, and is well tolerated. The hypotension can be due to peripheral vasodilatation (renin blockade), or a decrease in cardiac output, (β1-receptor blockade). Hypotension that is sudden in onset is usually a reflection of a decrease in cardiac output and is an ominous sign.
Membrane-Stabilizing Effect
Excessive doses of β-blockers can exert a membrane-stabilizing effect that is independent of the β-Receptor blockade. The principal consequence of this membrane-stabilizing effect is prolonged atrioventricular (AV) conduction, which can progress to complete heart block (27).
Neurotoxicity
Most β-blockers are lipophilic, and have a tendency to accumulate in lipid-rich tissues like the central nervous system. As a result, β-blocker overdose is often accompanied by lethargy, depressed consciousness, and generalized seizures. The latter manifestation is more prevalent than suspected, and has been reported in 60% of overdoses with propranolol (28). Like the prolonged AV conduction, the neurological manifestations are not the result of β-Receptor blockade, and may be related to the membrane-stabilizing effect.
Glucagon
Glucagon is a regulatory hormone that acts in opposition to insulin by stimulating glycogen breakdown to raise blood glucose levels. In a seemingly unrelated role, glucagon antagonizes the cardiac depression produced by β-Receptor antagonists.
Mechanism of Action
The illustration in Figure 54.4 shows how glucagon can correct the cardiac depression caused by β-blockers and calcium channel blockers. The glucagon receptor and β-Receptor are linked to the adenyl cyclase en-zyme on the inner surface of the cell membrane. Activation of each receptor-enzyme complex results in the hydrolysis of adenosine triphosphate (ATP) and the formation of cyclic adenosine monophosphate (cyclic AMP). The cyclic AMP then activates a protein kinase that promotes the inward movement of calcium through the cell membrane. The influx of calcium promotes interactions between contractile proteins to enhance the strength of cardiac contraction.
This sequence of reactions just described is responsible for the positive inotropic and chronotropic effects of β-Receptor stimulation. Since the same reactions occur with activation of the glucagon receptor, glucagon has positive inotropic and chronotropic effects that are equivalent to β-Receptor agonists. More importantly, the parallel orientation of the gluca-gon receptor and β-Receptor allows glucagon to retain its cardiostimulatory effects when β-Receptors are quiescent. This is the basis for gluca-gon’s role as an antidote for β-blocker toxicity.
Clinical Use
Glucagon is indicated for the treatment of hypotension and symptomatic bradycardia associated with toxic exposure to β-blockers. When used in the appropriate doses, glucagon will elicit a favorable response in 90% of patients (29). Glucagon is not indicated for reversing the prolonged AV conduction or neurological abnormalities in β-blocker overdoses because these effects are not mediated by β-Receptor blockade.
CALCIUM ANTAGONIST TOXICITY: Glucagon is also capable of antagonizing the effects of calcium channel blockers (30), as illustrated in Figure 54.4. However, glucagon is less effective in reversing the cardiac depression precipitated by calcium channel blocker overdoses. This is not unexpected, because glucagon and calcium blockers act at different sites in the pathways that modulate cardiac contractile strength.
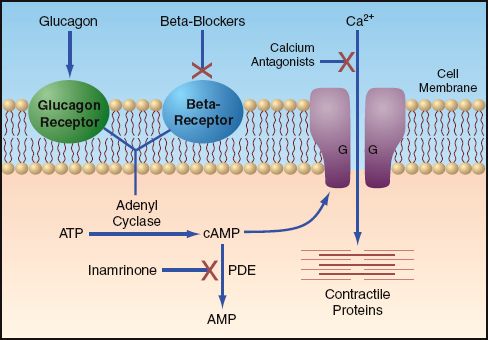
FIGURE 54.4 Mechanisms of drug-induced alterations in cardiac contractile strength. See text for explanation. Abbreviations: ATP=adenosine triphosphate; cAMP=cyclic adenosine monophosphate; PDE=phosphodiesterase; AMP=adenosine monophosphate.
Dosing Regimen
Dosing recommendations for glucagon are presented in Table 54.2. The effective dose of glucagon can vary in individual patients, but a bolus dose of 3–5 mg IV should be effective in most adults (28,29). The initial dose is 3 mg (or 0.05 mg/kg) as an IV bolus. The response to glucagon is usually evident within 3 minutes (30). If the response is not satisfactory, a second IV bolus dose of 5 mg (or 0.07 mg/kg) can be administered. The effects of glucagon can be short-lived (5 minutes), and so a favorable response should be followed by a continuous infusion (5 mg/hr). The chronotropic response to glucagon is optimal when the plasma ionized calcium is normal (31).
Table 54.2 Glucagon as an Antidote

Adverse Effects
Nausea and vomiting are common at glucagon doses above 5 mg/hr. Mild hyperglycemia is common, and is the result of glucagon’s actions to stimulate glycogenolysis. The insulin response to the hyperglycemia can drive potassium into cells and promote hypokalemia. Finally, glucagon stimulates catecholamine release from the adrenal medulla, and this can cause unwanted increases in blood pressure in patients with hypertension.
Phosphodiesterase Inhibitors
Phosphodiesterase inhibitors (e.g., inamrinone, milrinone) augment cardiac contractile strength by inhibiting the breakdown of cyclic AMP, as illustrated in Figure 54.4. These agents can increase cardiac output in the setting of β-blockade (32), and they should add to the increase in cyclic AMP produced by glucagon. However, it is unclear if phosphodiesterase inhibitors add to the effectiveness of glucagon in β-blocker toxicity. Because these drugs are vasodilators, and can produce unwanted decreases in blood pressure, they are generally reserved for cases of β-blocker toxicity that are resistant, or refractory, to glucagon.
OPIOIDS
Opiates are implicated in 75% of fatal drug overdoses in the United States (2a), and the problem is likely to grow rather than shrink. The adverse effects of opioids are described in Chapter 51; the following description focuses on the use of the narcotic antagonist, naloxone.
Clinical Features
The classic description of an opiate overdose is a patient who presents with stupor, pinpoint pupils, and slow breathing (bradypnea). However, these clinical findings are either absent or nonspecific, and it is not possible to identify an opiate overdose based on the clinical presentation or physical examination (33). The response to the narcotic antagonist, naloxone, is probably the most reliable method of identifying an opioid overdose.
Naloxone
Naloxone is a pure opioid antagonist; i.e., it binds to endogenous opioid receptors, but does not elicit any agonist responses. It is most effective in blocking mu receptors (primarily responsible for analgesia, euphoria, and respiratory depression) and less effective in blocking kappa receptors and delta receptors (32,33).
Dosing Regimen
Naloxone is usually given as an IV bolus, and the effect is apparent within 3 minutes. Alternate routes of delivery include intramuscular injection (onset in 15 minutes), intraosseous injection, intralingual injection, and endotracheal instillation (36). Reversing the sedative effects of opioids usually requires smaller doses of naloxone than reversing the respiratory depression.
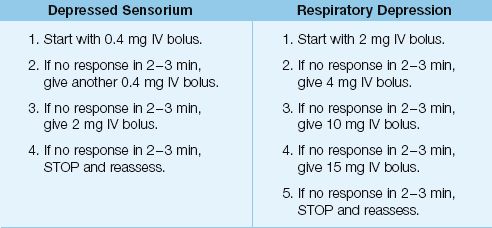
DEPRESSED SENSORIUM: For patients with a depressed sensorium but no respiratory depression, the initial dose of naloxone should be 0.4 mg IV push. This can be repeated in 2 minutes, if necessary. A total dose of 0.8 mg should be effective if the mental status changes are caused by an opioid derivative (24).
Respiratory Depression: For patients who have evidence of respiratory depression (e.g., hypercapnia, respiratory rate <12 breaths/min), the initial dose of naloxone should be 2 mg IV push. If there is no response in 2–3 minutes, double the initial dose (i.e., give 4 mg IV bolus). If further doses are needed, each successive dose should be increased as shown in Table 54.3, until the dose reaches 15 mg (33). Opioid overdose is unlikely if there is no response to a naloxone dose of 15 mg.
The effects of naloxone last about 60–90 minutes, which is less than the duration of action of most opioids. Therefore, a favorable response to naloxone should be followed by repeat doses at one-hour intervals, or by a continuous infusion. For a continuous naloxone infusion, the hourly dose of naloxone should be two-thirds of the effective bolus dose (diluted in 250 or 500 mL of isotonic saline and infused over 6 hours) (37). To achieve steady-state drug levels in the early infusion period, a second bolus of naloxone (at one-half the original bolus dose) is given 30 minutes after the infusion is started. The duration of treatment varies (ac-cording to the drug and the dose ingested), but averages 10 hours (24).
Table 54.3 Naloxone Dosing Regimens
Empiric Naloxone
Empiric therapy with naloxone (0.2–8 mg IV bolus) has been used in patients with altered mentation to identify occult cases of opioid overdose. However, this practice is effective in fewer than 5% of patients with altered mental status of unknown etiology (38). An alternative approach has been proposed where empiric naloxone is indicated only for patients with pinpoint pupils and circumstantial evidence of opioid abuse (e.g., needle tracks) (24,38). When naloxone is used in this manner, a favorable response (indicating an opioid overdose) is expected in about 90% of patients (38).
Adverse Reactions
Naloxone has few undesirable effects. The most common adverse reaction is the opioid withdrawal syndrome (anxiety, abdominal cramps, vomiting, and piloerection). There are case reports of acute pulmonary edema (most in the early postoperative period) and generalized seizures following naloxone administration (24), but these are rare occurrences.
SALICYLATES
Despite a steady decline in prevalence, salicylate intoxication is the 14th leading cause of drug-induced deaths in the United States (39).
Clinical Features
Ingestion of 10–30 grams of aspirin (150 mg/kg) can have fatal consequences. Once ingested, acetylsalicylic acid (aspirin) is promptly converted to salicylic acid, which is the active form of the drug. Salicylic acid is readily absorbed from the upper GI tract, and metabolism takes place in the liver. The hallmark of salicylate intoxication is the combination of a respiratory alkalosis and a metabolic acidosis.
Respiratory Alkalosis
Within hours after a toxic ingestion of aspirin, there is an increase in respiratory rate and tidal volume. This is the result of direct stimulation of brainstem respiratory neurons by salicylic acid, and the subsequent increase in minute ventilation results in a decrease in arterial PCO2 (i.e., acute respiratory alkalosis).
Metabolic Acidosis
Salicylic acid is a weak acid that does not readily dissociate, and thus does not produce a metabolic acidosis. However, salicylic acid activates proteins in the mitochondria that uncouple oxidative phosphorylation, and this results in a marked increase in the anaerobic production of lactic acid, and this is the principal source of the metabolic acidosis in salicylate intoxication. The mixed metabolic acidosis-respiratory alkalosis produces an arterial blood gas with a low PCO2, a low bicarbonate, and a normal pH (see Rule 3 on page 593). As the lactic acidosis progresses, the serum pH will eventually fall, which is a poor prognostic sign (40).
Other Features
Clinical features in the early stages of salicylate toxicity include nausea, vomiting, tinnitus, and agitation. Progression of the toxidrome is associated with neurologic changes (delirium, seizures, and progression to coma), fever (from the uncoupled oxidative phosphorylation), and acute respiratory distress syndrome (ARDS).
Diagnosis
The plasma salicylate level is used to confirm or exclude the diagnosis of salicylate toxicity. The therapeutic range of salicylates in plasma is 10–30 mg/L (0.7–2.2 mmol/L), and levels above 40 mg/L (2.9 mmol/L) are considered toxic (40). Plasma salicylate levels are usually elevated within 4–6 hours after a toxic ingestion.
Management
Management of salicylate toxicity includes general supportive care (i.e., fluids, vasopressors, and mechanical ventilation), if necessary. Multiple-dose activated charcoal is recommended, if it can be started within 2–3 hours of drug ingestion. The dosing regimen is 25 grams orally every 2 hours for 3 doses.
Alkalinization
Alkalinization of the urine to enhance salicylate excretion is the cornerstone of management for salicylate intoxication. Salicylic acid has a pK of 3 (the pK is the pH at which 50% of the acid is dissociated), which means that the acid will dissociate more readily as the pH rises. An alkaline pH in the urine will promote dissociation of salicylic acid in the renal tubules, and this essentially “traps” the salicylic acid in the renal tubular lumen, where it can be excreted in the urine. Bicarbonate infusions are used to raise the urine pH using the following regimen, which is summarized in Table 54.4.
Table 54.4 Protocol for Alkalinization of the Urine

REGIMEN: Start IV bicarbonate therapy with 1–2 mEq/kg as an IV bolus, and follow with a continuous bicarbonate infusion. To create the bicarbonate infusate, add 3 ampules of 40% sodium bicarbonate (43 mEq/amp) to 1 liter of D5W (129 mEq/L), and infuse this solution at 2–3 mL/kg/hr (40). The urine pH should be maintained at ≥7.5 (41). Treatment continues until the plasma salicylate levels fall below the toxic range.
HYPOKALEMIA: Bicarbonate infusions will lower the serum potassium (intracellular shift), and hypokalemia hampers the ability to alkalinize the urine. This is explained by the K+ H+ exchanger in the distal renal tubules, which releases H+ into the tubular fluid as K+ is reabsorbed. Since K+ reabsorption is increased in the setting of hypokalemia, the extra H+ released into the tubular fluid will hamper attempts to alkalinize the urine. Potassium should be added to the bicarbonate solution (40 mEq/L) to reduce the risk of hypokalemia.
Hemodialysis
Hemodialysis is the most effective method of clearing salicylates from the body (42). The indications for hemodialysis include a serum salicylate level >100 mg/L, the presence of renal failure or ARDS, or progression of the toxidrome despite alkalinization therapy (39).
A FINAL WORD
Regulating Acetaminophen
If any over-the-counter drug deserves to be regulated, it’s acetaminophen. Not because it is the leading cause of acute liver failure in the United States (which should be enough of a reason), but because half of the overdoses are unintentional, indicating a general lack of awareness of the toxic potential of acetaminophen (which can be fatal). The FDA has called for stronger warnings on drug packaging, but who reads that small print? Requiring a prescription that specifies how the drug should be used is the best way to ensure that acetaminophen is used safely in the general population.
Acetaminophen gained its popularity in the 1970s because of concerns about the toxicity of aspirin, and it looks like we moved from the frying pan into the fire.
REFERENCES
Introduction
1. Centers for Disease Control and Prevention. Vital signs: overdoses of prescribed opioid pain relievers—United States, 1999–2008. MMWR 2011; 60:1487–1492.
2. Centers for Disease Control and Prevention. National Vital Statistics System. 2010 Multiple Cause of Death File. Hyattsville, MD: US Department of Health and Human Services, Centers for Disease Control and Prevention; 2012.
2a. Jones CM, Mack KA, Paulozzi LJ. Pharmaceutical overdose deaths, United States, 2010. JAMA 2013; 309:657–659.
Acetaminophen
3. Hodgman M, Garrard AR. A review of acetaminophen poisoning. Crit Care Clin 2012; 28:499–516.
4. Larson AM, Polson J, Fontana RJ, et al. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
5. Kuehn B. FDA focuses on drugs and liver damage. JAMA 2009; 302:369–370.
6. Hendrickson RG, Bizovi KE. Acetaminophen. In: Flomenbaum NE, et al., eds. Goldfrank’s Toxicologic Emergencies. 8th ed. New York: McGraw-Hill, 2006; 523–543.
7. Rumack BH. Acetaminophen hepatotoxicity: the first 35 years. J Toxicol Clin Toxicol 2002; 40:3–20.
N-Acetylcysteine
8. Holdiness MR. Clinical pharmacokinetics of N-acetylcysteine. Clin Pharma-cokinet 1991; 20:123–134.
9. Rumack BH, Peterson RC, Koch GG, et al. Acetaminophen overdose. 662 cases with evaluation of oral acetylcysteine treatment. Arch Int Med 1981; 141:380–385.
10. Harrison PM, Keays R, Bray GP, et al. Improved outcome of paracetamol-induced fulminant hepatic failure by late administration of acetylcysteine. Lancet 1990; 335:1572–1573.
11. Cumberland Pharmaceuticals. Acetadote Package Insert. 2006.
12. Smilkstein MJ, Knapp GL, Kulig KW, et al. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 1988; 319:1557–1562.
13. Howland MA. N-Acetylcysteine. In: Flomenbaum NE, et al., eds. Goldfrank’s Toxicologic Emergencies. 8th ed. New York: McGraw-Hill, 2006; 544–549.
14. Buckley NA, Whyte IM, O’Connell DL, et al. Oral or intravenous N-acetylcysteine: which is the treatment of choice for acetaminophen (paracetamol) poisoning? J Toxicol Clin Toxicol 1999; 37:759–767.
15. Appelboam AV, Dargan PI, Knighton J. Fatal anaphylactoid reaction to N-acetylcysteine: caution in patients with asthma. Emerg Med J 2002; 19:594–595.
16. Miller LF, Rumack BH. Clinical safety of high oral doses of acetylcysteine. Semin Oncol 1983; 10:76–85.
17. Spiller HA, Krenzelok EP, Grande GA, et al. A prospective evaluation of the effect of activated charcoal before oral N-acetylcysteine in acetaminophen overdose. Ann Emerg Med 1994; 23:519–523.
Benzodiazepines
18. Gaudreault P, Guay J, Thivierge RL, Verdy I. Benzodiazepine poisoning. Drug Saf 1991; 6:247–265.
19. Wu AH, McCay C, Broussard LA, et al. National Academy of Clinical Biochemistry laboratory medicine practice guidelines: Recommendations for the use of laboratory tests to support poisoned patients who present to the emergency department. Clin Chem 2003; 49:357–379.
Flumazenil
20. Howland MA. Flumazenil. In: Flomenbaum NE, et al., eds. Goldfrank’s Toxicologic Emergencies. 8th ed. New York: McGraw-Hill, 2006; 1112–1117.
21. Shalansky SJ, Naumann TL, Englander FA. Effect of flumazenil on benzodiazepine-induced respiratory depression. Clin Pharm 1993; 12:483–487.
22. Roche Laboratories. Romazicon (flumazenil) package insert. 2004.
23. Bodenham A, Park GR. Reversal of prolonged sedation using flumazenil in critically ill patients. Anaesthesia 1989; 44:603–605.
24. Doyon S, Roberts JR. Reappraisal of the “coma cocktail”. Dextrose, flumazenil, naloxone, and thiamine. Emerg Med Clin North Am 1994; 12:301–316.
25. Haverkos GP, DiSalvo RP, Imhoff TE. Fatal seizures after flumazenil administration in a patient with mixed overdose. Ann Pharmacother 1994; 28:1347–1349.
ß-Receptor Antagonists
26. Newton CR, Delgado JH, Gomez HF. Calcium and beta receptor antagonist overdose: a review and update of pharmacological principles and management. Semin Respir Crit Care Med 2002; 23:19–25.
27. Henry JA, Cassidy SL. Membrane stabilising activity: a major cause of fatal poisoning. Lancet 1986; 1:1414–1417.
28. Weinstein RS. Recognition and management of poisoning with beta-adrenergic blocking agents. Ann Emerg Med 1984; 13:1123–1131.
29. Kerns W, 2nd, Kline J, Ford MD. Beta-blocker and calcium channel blocker toxicity. Emerg Med Clin North Am 1994; 12:365–390.
Glucagon
30. Howland MA. Glucagon. In: Flomenbaum NE, et al., eds. Goldfrank’s Toxicologic Emergencies. 8th ed. New York: McGraw-Hill, 2006; 942–945.
31. Chernow B, Zaloga GP, Malcolm D, et al. Glucagon’s chronotropic action is calcium dependent. J Pharmacol Exp Ther 1987; 241:833–837.
32. Travill CM, Pugh S, Noblr MI. The inotropic and hemodynamic effects of intravenous milrinone when reflex adrenergic stimulation is suppressed by beta adrenergic blockade. Clin Ther 1994; 16:783–792.
Opioids and Naloxone
33. Boyer EW. Management of opioid analgesic overdose. N Engl J Med 2012; 367:146–155.
34. Handal KA, Schauben JL, Salamone FR. Naloxone. Ann Emerg Med 1983; 12:438–445.
35. Howland MA. Opioid Antagonists. In: Flomenbaum NE, et al., eds. Goldfrank’s Toxicologic Emergencies. 8th ed. New York: McGraw-Hill, 2006; 614–619.
36. Naloxone hydrochloride. In: McEvoy GK ed. AHFS Drug Information, 2012. Bethesda: American Society of Hospital Systems Pharmacists, 2012:2236–2239.
37. Goldfrank L, Weisman RS, Errick JK, et al. A dosing nomogram for continuous infusion intravenous naloxone. Ann Emerg Med 1986; 15:566–570.
38. Hoffman JR, Schriger DL, Luo JS. The empiric use of naloxone in patients with altered mental status: a reappraisal. Ann Emerg Med 1991; 20:246–252.
Salicylates
39. Bronstein AC, Spyker DA, Cantilena LR, et al. 2011 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS):29th Annual Report. Clin Toxicol 2012; 50:911–1164.
40. O’Malley GF. Emergency department management of the salicylate-poisoned patient. Emerg Med Clin N Am 2007; 25:333–346.
41. Proudfoot AT, Krenzelok EP, Vale JA. Position paper on urine alkalinization. J Toxicol Clin Toxicol 2004; 42:1–26.
42. Fertel BS, Nelson LS, Goldfarb DS. The underutilization of hemodialysis in patients with salicylate poisoning. Kidney Int 2009; 75:1349–1353.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


