FIGURE 51.1 Percentage of housestaff physicians and ICU nurses who answered incorrectly when asked if diazepam (Valium) relieved pain.
Pain in Critically Ill Patients
Surveys indicate that 50–80% of ICU patients experience pain and discomfort while in the ICU (2,6,7). Surprisingly, the prevalence of pain is the same in surgical ICUs and medical ICUs (10).
Hypernociception
Critically ill patients experience pain more readily than healthy subjects (hypernociception). For example, the most painful experiences for ICU patients are endotracheal suctioning, and being turned in bed (8). In addition, 30–50% of critically ill patients experience pain while at rest, without a noxious stimulus (8,10). This type of pain typically involves the back and lower extremities.
The heightened pain experience in critically ill patients is attributed to immobility and systemic inflammation. (The rest pain experienced by ICU patients is similar to the myalgias experienced during a systemic infection.) Failure to recognize this heightened nociceptive state is a major source of inadequate pain relief in the ICU. Frequent pain assessments using pain intensity scales (described next) can help to correct problems with inadequate pain control in the ICU (3).
Monitoring Pain
Pain is defined as “an unpleasant sensory or emotional experience” (11), which highlights the subjective nature of pain. The pain sensation can be described in terms of intensity, duration, location, and quality, but pain intensity is the parameter most often monitored because it reflects the “unpleasantness” of pain.
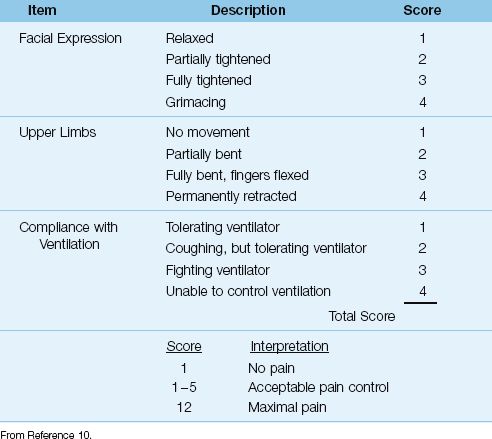
Table 51.1 The Behavioral Pain Scale
Pain Intensity Scales
Pain intensity scales are used to determine the need for, and evaluate the effectiveness of, analgesic therapy. There are 6 different pain intensity scales, but only a few are needed to assess pain intensity in most ICU patients.
1. When patients can reliably self-report pain, the Numerical Ranking Scale can be used for pain assessment (12). This is a ruler with 10 equally spaced divider markings, numbered 1 (no pain) to 10 (maximum pain). The patient points to one of the numbered markings to indicate the severity of pain. A score of 3 or less is considered adequate pain control.
2. When patients are sedated and on a ventilator, the Behavioral Pain Scale (BPS) shown in Table 51.1 is recommended for pain assessments (3). This scale evaluates pain intensity by elicited behaviors (i.e., facial expression, arm flexion, and tolerance of mechanical ventilation) (11). Scores can range from 3 (no pain) to 12 (maximum pain). A score of 5 or less represents adequate pain control (3).
Vital Signs
Vital signs (e.g., heart rate) show a poor correlation with patients’ reports of pain intensity (the gold standard), and they can remain unchanged in the presence of pain (13). As a result, vital signs are not recommended for pain assessments (3).
OPIOID ANALGESIA
The natural chemical derivatives of opium are called opiates. Opiates and other substances that produce their effects by stimulating discrete opioid receptors in the central nervous system are called opioids. Stimulation of opioid receptors produces a variety of effects, including analgesia, sedation, euphoria, pupillary constriction, respiratory depression, bradycardia, constipation, nausea, vomiting, urinary retention, and pruritis (14). The term narcotic refers to a general class of drugs that blunt sensation and depress consciousness (i.e., narcotize).
Opioids are the most frequently used drugs for pain relief in the ICU (3,8), and are given intravenously as intermittent bolus doses or continuous infusions. Opioids also produce mild sedation but, unlike benzodiazepines, they have no amnestic effects (15).
Drugs and Dosing Regimens
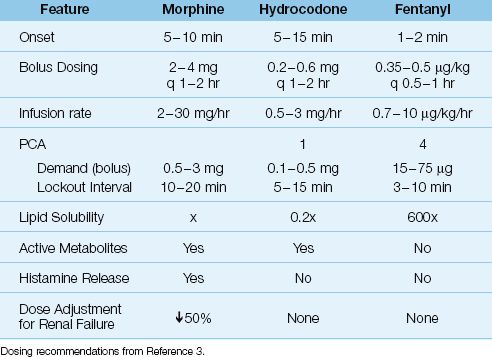
The opioids used most often in the ICU are morphine, fentanyl, and hydromorphone. The recommended intravenous doses for each drug are shown in Table 51.2. It is important to emphasize that opioid dose requirements can vary widely in individual patients, and that the effective dose of an opioid is determined by each patient’s response, not by the recommended dose range of the drug.
Fentanyl
Fentanyl has replaced morphine as the most popular opioid analgesic in ICUs (8). The advantages of fentanyl over morphine include a more rapid onset of action (because fentanyl is 600 times more lipid soluble than morphine), less risk of hypotension (because fentanyl does not promote histamine release) (16), and the absence of active metabolites. The relative lack of adverse hemodynamic effects is a major source of fentanyl’s appeal in critically ill patients.
Morphine
Opioids are metabolized primarily in the liver, and the metabolites are excreted in the urine. Morphine has several active metabolites that can accumulate in renal failure. One metabolite (morphine 3-glucuronide) can produce central nervous system excitation with myoclonus and seizures (17), while another metabolite (morphine-6-glucuronide) has more potent analgesic effects than the parent drug (14). To avoid accumulation of these metabolites, the maintenance dose of morphine should be reduced by 50% in patients with renal failure (18). Fentanyl does not have active metabolites, and dose not require dose adjustments in renal failure.
Morphine also promotes the release of histamine, and this can produce systemic vasodilation and a decrease in blood pressure (14). Hypotension can occur, and is typically seen in patients with a hyperadrenergic state and increased peripheral vascular tone (4). Morphine-induced histamine release does not promote bronchoconstriction, and morphine doses of 1.5 mg/kg have been given to asthma patients without adverse consequences (19).
Hydromorphone
Hydromorphone is a morphine derivative that may produce more effective analgesia (according to a recent meta-analysis) (20). However, other than the lack of a need for dose alterations in renal failure, hydromorphone has no apparent clinical advantage over morphine in critically ill patients.
Table 51.2 Commonly Used Intravenous Opioids
Remifentanil
Remifentanil is an ultra-short acting opioid that is given by continuous intravenous infusion, using the dosing regimen shown below.
Dosing Regimen: 1.5 µg/kg as a loading dose, followed by a continuous infusion at 0.5–15 µg/kg/hr (3).
Analgesic effects are lost within 10 minutes after stopping the drug infusion. The short duration of action is a reflection of drug metabolism; i.e., remifentanil is broken down by nonspecific esterases in plasma. Because drug metabolism does not take place in the liver or kidneys, dose adjustments are not necessary in renal or hepatic failure.
Remifentanil’s short duration of action is most advantageous in conditions that require frequent evaluations of cerebral function (e.g., traumatic head injuries). The abrupt cessation of opioid activity can precipitate acute opioid withdrawal (5), which can be prevented by combining remifentanil with a longer-acting opioid.
Meperidine
Meperidine (Demerol, Pethidine) is an opioid analgesic that is no longer favored for pain control in the ICU because of the potential for neurotoxicity. Meperidine is metabolized in the liver to normeperidine, a metabolite that is slowly excreted by the kidneys (elimination half-life is 15–40 hours) (21). Accumulation of normeperidine can produce central nervous system excitation, with agitation, myoclonus, delirium, and generalized seizures (21). Since renal dysfunction is prevalent in ICU patients, there is a high risk for accumulation of the neurotoxic metabolite of meperidine in ICU patients.
Patient-Controlled Analgesia
For patients who are awake and capable of drug self-administration, patient-controlled analgesia (PCA) can be an effective method of pain control, and may be superior to intermittent opioid dosing. The PCA method uses an electronic infusion pump that can be activated by the patient. When pain is sensed, the patient presses a button connected to the pump to receive a small intravenous bolus of drug. After each bolus, the pump is disabled for a mandatory time period called the “lockout interval,” to prevent overdosing. The opioid dosing regimens for PCA are shown in Table 51.2. The minimum lockout interval is determined by the time required to achieve peak drug effect (22). When writing orders for PCA, you must specify the initial loading dose (if any), the lockout interval, and the repeat bolus dose. PCA can be used alone or in conjunction with a low-dose opioid infusion.
Adverse Effects of Opioids
Respiratory Depression
Opioids produce a centrally mediated, dose-dependent decrease in respiratory rate and tidal volume (23,24), but respiratory depression and hypoxemia are uncommon when opioids are given in the usual doses (25). Opioid doses that impair arousal also impair ventilation and produce hypercapnia (23). Patients with sleep apnea syndrome or chronic hypercapnia are particularly prone to respiratory depression from opioids.
Cardiovascular Effects
Opioid analgesia is often accompanied by decreases in blood pressure and heart rate, which are the result of decreased sympathetic activity and in-creased parasympathetic activity. These effects are usually mild and well-tolerated, at least in the supine position (26). Decreases in blood pressure can be pronounced in patients with hypovolemia or heart failure (where there is an increased baseline sympathetic tone), or when opioids are given in combination with benzodiazepines (27). Opioid-induced hypo-tension is rarely a threat to tissue perfusion, and the blood pressure re-sponds to intravenous fluids or small bolus doses of vasopressors.
Intestinal Motility
Opioids are well known for their ability to depress bowel motility via activation of opioid receptors in the GI tract. This is a source of troublesome constipation in cancer patients. In critically ill patients, impaired GI motility can promote reflux of enteral tube feedings into the oropharynx, creating a risk for aspiration pneumonia. Opioid-induced bowel hypomotility can be partially reversed with enteral naloxone (8 mg every 6 hours) without affecting opioid analgesia. In one small study (28), this approach resulted in fewer cases of aspiration pneumonia.
Nausea and Vomiting
Opioids can promote vomiting via stimulation of the chemoreceptor trigger zone in the lower brainstem (23). All opioids are equivalent in their ability to promote vomiting, but substituting one opioid for another sometimes resolves the problem.
NON-OPIOID ANALGESIA
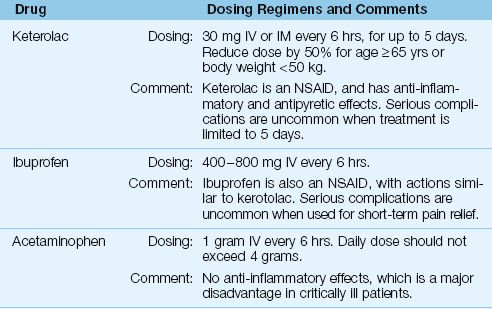
The list of intravenous non-opioid analgesics is a very short one, and includes only three drugs: acetaminophen, ketorolac, and ibuprofen. These drugs are used primarily for pain control in the early postoperative period. They can be used alone for mild pain, but are used in combination with an opioid analgesic for moderate-to-severe pain. The goal of combination analgesic therapy is to reduce the opioid dose (“opioid sparing” effect) and hence reduce the risk of an opioid-related adverse effect. The intravenous dosing regimens for the non-opioid analgesics are presented in Table 51.3.
Ketorolac
Ketorolac is a nonsteroidal anti-inflammatory drug (NSAID) that was introduced in 1990 as the first parenteral analgesic agent for postoperative pain that does not produce respiratory depression (29). Ketorolac has a proven opioid sparing effect, and the dose of opioid analgesics can often be reduced by 25–50% (30).
Dosing Regimen
Ketorolac can be given by intravenous (IV) or intramuscular (IM) injection. The recommended dosing regimen for moderate-to-severe pain in adults is 30 mg IV or IM every 6 hrs, for up to 5 days (31). A dose reduction of 50% is recommended for elderly patients (age ≥65 yrs), and for patients with a body weight <50 kg. IM injection of ketorolac can produce hematomas (32), so IV bolus injection may be preferred.
Risk of Adversity
The beneficial actions of ketorolac and other NSAIDS are attributed to inhibition of prostaglandin production, but this also creates a risk for adverse effects, particularly gastric mucosal injury, upper GI hemorrhage, and impairment of renal function (31). These adverse effects are typically associated with excessive dosing or prolonged exposure to NSAIDS, and they are uncommon when ketorolac is given in the recommended doses, and the treatment period is limited to 5 days (32–34).
Table 51.3 Intravenous Non-Opioid Analgesia
Ibuprofen
Ibuprofen is very similar to ketorolac because (a) it is an NSAID that can be given intravenously, (b) it has an opioid sparing effect, and (c) it is safe when used for short-term pain control (35). The intravenous dose of ibuprofen is 400-800 mg IV every 6 hrs, with a maximum daily dose of 3.2 grams (35). Unlike ketorolac, the treatment period for ibuprofen has no recommended time limit. Clinical trials of IV ibuprofen typically employ a treatment period of 24–48 hrs, and serious complications are uncommon over that time period.
Acetaminophen
Acetaminophen was approved for intravenous use in 2010, and is intended for the short-term treatment of pain and fever in postoperative patients who are unable to receive acetaminophen via the oral or rectal routes (36). The recommended dose for IV acetaminophen is 1 gram every 6 hrs, with a maximum allowable dose of 4 grams daily (to prevent acetaminophen hepatotoxicity) (36). This dosing regimen has a documented opiate sparing effect in postoperative patients (37).
Disadvantages
Acetaminophen has no anti-inflammatory activity, which is a major disadvantage in critically ill patients. Furthermore, although the daily dose limitation of 4 grams is intended to avoid acetaminophen hepatotoxicity, the toxic dose of acetaminophen has not been evaluated in critically ill patients. For these reasons, IV acetaminophen is not the optimal choice for opiate-sparing analgesia in critically ill patients. (For a description of acetaminophen hepatotoxicity, see Chapter 54.)
Neuropathic Pain
Non-opioid analgesia is usually required for neuropathic pain (e.g., from diabetic neuropathy), and the recommended drugs for this type of pain are gabapentin and carbamazepine (3). Both drugs must be given enterally. Effective drug doses vary in individual patients, but typical doses are 600 mg every 8 hrs for gabapentin, and 100 mg every 6 hours for carbamazepine (oral suspension).
ANXIETY IN THE ICU
Anxiety and related disorders (agitation and delirium) are observed in as many as 85% of patients in the ICU (38). These disorders can be defined as follows.
1. Anxiety is characterized by exaggerated feelings of fear or apprehension that are sustained by internal mechanisms more than external events.
2. Agitation is a state of anxiety that is accompanied by increased motor activity.
3. Delirium is an acute confusional state that may, or may not, have agitation as a component. Although delirium is often equated with agitation, there is a hypoactive form of delirium that is characterized by lethargy. (Delirium is described in more detail in Chapter 44.)
The common denominator in these disorders is the absence of a sense of well-being.
Sedation
Sedation is the process of relieving anxiety and establishing a state of calm. This process includes general supportive measures (like frequent communication with patients and families), and drug therapy. The drugs used most often for sedation in ICUs are midazolam and propofol (3).
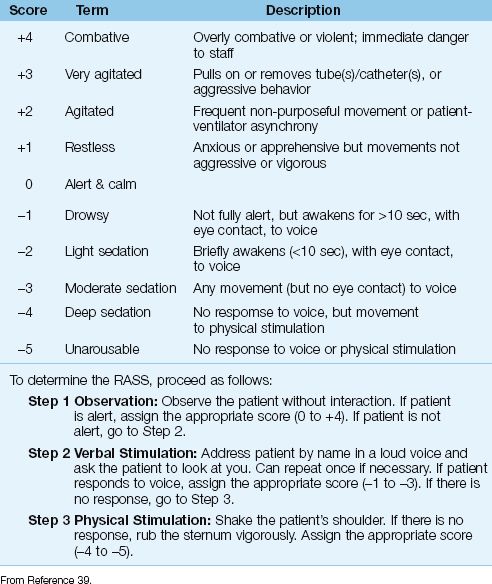
Table 51.4 Richmond Agitation Sedation Scale (RASS)
Monitoring Sedation
The routine use of sedation scales is instrumental in achieving effective sedation (3). The sedation scales that are most reliable in ICU patients are the Sedation-Agitation Scale (SAS) and the Richmond Agitation-Sedation Scale (RASS) (3), and the latter scale is shown in Table 51.4 (39). The RASS includes 4 possible scores for progressive agitation (+1 to +4) and 5 possible scores for progressive sedation (-1 to -5). The optimal RASS score is zero (alert and calm). The added advantage of RASS is the ability to monitor serial changes in a patient’s mental state (40). This latter feature allows the RASS score to be used as the end-point of sedative drug therapy. (Sedative drug infusions can be titrated to achieve a RASS score of –1 to –2, which represents light sedation.)
BENZODIAZEPINES
Benzodiazepines are currently the most popular drugs for sedation in ICUs (3,8), but they are gradually losing ground to other sedatives because of problems with drug accumulation and excessive sedation.
Drug Profiles
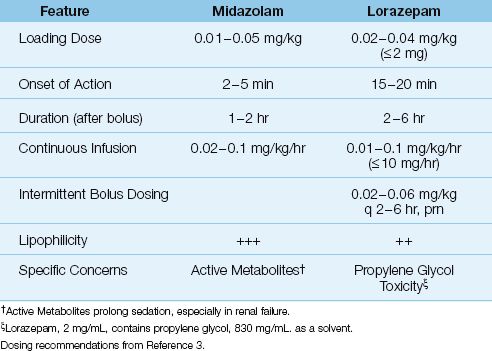
Two benzodiazepines are used for sedation in ICUs: midazolam and lorazepam. (Diazepam is no longer used because of excessive sedation with prolonged use.) Both drugs are given intravenously, and a brief profile of each drug is presented in Table 51.5.
Midazolam
Midazolam (Versed) is a rapid-acting drug by virtue of its high lipid solubility. Sedative effects are apparent within 1–2 minutes after an intravenous (IV) injection of midazolam, and this makes midazolam the preferred benzodiazepine for rapid sedation (e.g., for a severely agitated or combative patient). The avid uptake of midazolam into tissues also results in rapid clearance from the bloodstream, resulting in a short duration of action (41). Because of the short-lived effect (1–2 hrs), midazolam is given as a continuous IV infusion, preceded by a bolus loading dose. However, because the brief drug effect is due to avid drug uptake into tissues, rather than drug elimination from the body, a continuous infusion of midazolam will result in progressive drug accumulation in tissues. To avoid excessive sedation from drug accumulation, midazolam infusions should be limited to ≤48 hrs (4).
Lorazepam
Lorazepam (Ativan) is a longer-acting drug than midazolam, with effects lasting up to 6 hours after a single intravenous dose (3). Lorazepam can given by intermittent IV injections, or by continuous IV infusion. The intravenous preparation of lorazepam contains propylene glycol, a solvent used to increase drug solubility in plasma. This solvent has adverse effects (see later), which is why the lorazepam dosing recommendations in Table 51.5 have a maximum allowable dose (2 mg for bolus doses of lorazepam, and 10 mg/hr for continuous infusions).
Metabolism
Benzodiazepines are metabolized in the liver. Midazolam is metabolized by the cytochrome P450 enzyme system, and drugs that interfere with this enzyme system (e.g., diltiazem, erythromycin) can inhibit midazolam metabolism and potentiate it effects. Midazolam has one active metabolite, 1-hydroxymidazolam, that is cleared by the kidneys, so alterations in renal function can also influence midazolam sedation. Lorazepam is metabolized by glucuronidation, and has no active metabolites.
Table 51.5 Sedation with Intravenous Benzodiazepines
Advantages
The advantages of sedation with benzodiazepines include the following.
1. Benzodiazepines have a dose-dependent amnestic effect that is distinct from the sedative effect. The amnesia extends beyond the sedation period (antegrade amnesia), and this may be responsible for the surprising percentage (up to 40%) of patients who, after discharge from the ICU, have no recollection of events during their ICU stay (2,6,7). As mentioned earlier, this amnesia should be beneficial because it eliminates memories of stressful experiences.
2. Benzodiazepines have anticonvulsant effects (see Chapter 45), which is always a benefit in critically ill patients.
3. Benzodiazepines are the sedatives of choice for drug withdrawal syndromes, including alcohol, opiate, and benzodiazepine withdrawal.
Disadvantages
The major disadvantages of sedation with benzodiazepines are (a) drug accumulation with prolonged sedation, and (b) the apparent tendency for benzodiazepines to promote delirium.
Prolonged Sedation
Maintaining sedation in ICU patients is often a long-term affair, especially in ventilator-dependent patients, and both midazolam and lorazepam accumulate in tissues with prolonged use. This produces deeper levels of sedation, and prolongs the time for awakening when the drugs are discontinued. This can result in delays in weaning patients from mechanical ventilation, and longer ICU stays (3,4). Prolonged sedation is more of a problem with midazolam, because of its greater lipid solubility, and the accumulation of its active metabolite. In one study of sedation in ICU patients, the time to emerge from sedation was 1,815 min (30.2 hours) for midazolam, versus 261 min (4.4 hr) for lorazepam (42).
SOLUTIONS: The following are some solutions to the problem of prolonged sedation with benzodiazepines:
1. Daily interruption of benzodiazepine infusions (until the patient awakens) curtails drug accumulation, and has been shown to shorten the duration of mechanical ventilation, and reduce the length of stay in the ICU (43). This is the accepted practice for limiting excessive sedation from benzodiazepines.
2. Titration of benzodiazepine infusions to maintain light levels of sedation, using routine monitoring with a sedation scale (SAS or RASS), has been proposed in the most recent guidelines on sedation in ICUs (3). This is a more logical solution to the problem than daily interruption of benzodiazepine infusions, and should be a standard practice.
3. The final solution to the problem is to avoid benzodiazepines for sedation, which is a current trend.
Delirium
The prevailing opinion is that the frequent appearance of delirium in ICU patients (described in Chapter 44) is at least partly due to the frequent use of benzodiazepines in ICU patients (3,4). Benzodiazepines produce their effects by binding to receptors for gamma-amino-butyric-acid (GABA), which are involved in the development of delirium (44), and sedation with drugs that do not involve GABA receptors is associated with fewer cases of delirium in ICU patients (see later). The role of benzodiazepines in promoting delirium will be a severe blow to their popularity if suitable alternatives are available.
Propylene Glycol Toxicity
Intravenous preparations of lorazepam contain propylene glycol (415 mg/mg lorazepam) to enhance drug solubility in plasma. Propylene glycol is converted to lactic acid in the liver, and excessive intake of propylene glycol can produce a toxidrome characterized by a metabolic (lactic) acidosis, delirium (with hallucination), hypotension, and (in severe cases) multiorgan failure. This toxidrome has been reported in 19–66% of patients receiving high-dose intravenous lorazepam for more than 2 days (45,46).
The maximum daily intake of propylene glycol that is considered safe is 25 mg/kg (47), or 17.5 g/day for a 70 kg adult. A lorazepam infusion at 2 mg/hr represents a daily intake of propylene glycol of 830 mg × 24 = 19.9 g/day, which exceeds the safe limit for a 70 kg adult. This highlights the risk of propylene glycol toxicity with lorazepam infusions that continue for 24 hrs or longer.
DIAGNOSIS: An unexplained metabolic acidosis during prolonged (>24 hr) infusions of lorazepam should prompt a measurement of the serum lactate levels, and an elevated lactate should raise suspicion of propylene glycol toxicity. Plasma levels of propylene glycol can be measured, but the results may not be immediately available. An elevated osmolal gap can suggest the diagnosis because propylene glycol will elevate the osmolal gap. (see page 656 for a description of the osmolal gap).
Withdrawal Syndrome
Abrupt termination of prolonged benzodiazepine infusions can produce a withdrawal syndrome, characterized by agitation, disorientation, hallucinations, and seizures (48). However, this does not appear to be a common occurrence.
OTHER SEDATIVES
Concerns about the delayed emergence from sedation with benzodiazepines has led to the increasing popularity of two sedatives that allow rapid arousal: propofol and dexmedetomidine.
Propofol
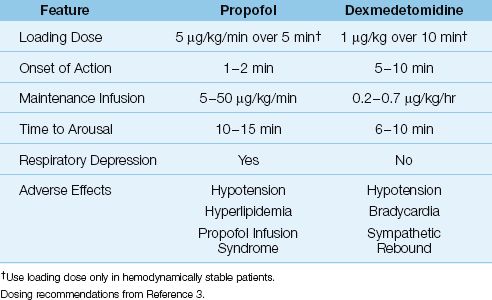
Propofol (Deprivan) is a powerful sedative that exerts its effects by binding to GABA receptors (similar to benzodiazepines, but different receptors). It is used for induction of general anesthesia, and has become popular in ICUs because arousal is rapid when the drug infusion is stopped. A profile of this drug is presented in Table 51.6.
Actions and Uses
Propofol has sedative and amnestic effects, but no analgesic effects (49). A single intravenous bolus of propofol produces sedation within 1–2 minutes, and the drug effect lasts 5–8 minutes (49). Because of the short duration of action, propofol is given as a continuous infusion. When the infusion is stopped, awakening occurs within 10–15 minutes, even with prolonged infusions (49). Loading doses of propofol can be used in pa-tients who are hemodynamically stable.
Propofol was originally intended for short-term sedation when rapid awakening is desired (e.g., during brief procedures), but it is being used for longer periods of time in ventilator-dependent patients, to avoid delays in weaning from ventilatory support. Propofol can be useful in neurosurgical patients and patients with head injuries because it reduces intracranial pressure (49), and the rapid arousal allows for frequent evaluations of mental status.
Preparation and Dosage
Propofol is highly lipophilic, and is suspended in a 10% lipid emulsion to enhance solubility in plasma. This lipid emulsion is almost identical to 10% Intralipid used in parenteral nutrition formulas, and it has a caloric density of 1 kcal/mL, which should be included as part of the daily caloric intake. Propofol dosing is based on ideal rather than actual body weight, and no dose adjustment is required for renal failure or moderate hepatic insufficiency (49). Loading doses are not advised in patients who are hemodynamically unstable (because of the risk of hypotension) (3).
Table 51.6 Sedation with Rapid-Arousal Drugs
Adverse Effects
Propofol is well known for producing respiratory depression and hypotension (50). Because of the risk of respiratory depression, propofol infusions are recommended only for ventilator-dependent patients. Hypotension is attributed to systemic vasodilatation (4), and can be profound in conditions like hypovolemia and heart failure, where blood pressure is maintained by systemic vasoconstriction. Anaphylactoid re-actions to propofol are uncommon but can be severe (49), and green urine is observed occasionally from harmless phenolic metabolites (49).
The lipid emulsion in propofol preparations can promote hypertriglyceridemia. The incidence is not known, but propofol infusions are the leading independent risk factor for hypertriglyceridemia in ICU patients (51). Monitoring triglyceride levels is often recommended during propofol infusions, but hypertriglyceridemia is common in ICU patients, and is not associated with adverse outcomes (51), so the merits of monitoring triglyceride levels are questionable.
Propofol Infusion Syndrome
Propofol infusion syndrome is a rare and poorly understood condition that is characterized by the abrupt onset of bradycardic heart failure, lactic acidosis, rhabdomyolysis, and acute renal failure (52). This syndrome almost always occurs during prolonged, high-dose propofol infusions (>4–6 mg/kg/hr for longer than 24–48 hrs) (53). The mortality rate is 30% (52). Avoiding propofol infusion rates above 5 mg/kg/hr for longer than 48 hrs is recommended to reduce the risk of this condition (53).
Dexmedetomidine
Dexmedetomidine is an alpha-2 receptor agonist that has sedative, amnestic, and mild analgesic effects, and does not depress ventilation. A brief profile of the drug is presented in Table 51.6. The most distinguishing feature of dexmedetomidine is the type of the sedation it produces, which is described next.
Cooperative Sedation
The sedation produced by dexmedetomidine is unique because arousal is maintained, despite deep levels of sedation. Patients can be aroused from sedation without discontinuing the drug infusion, and when awake, patients are able to communicate and follow commands. When arousal is no longer required, the patient is allowed to return to the prior state of sedation. This has been called cooperative sedation (5), and is similar to temporary awakening from sleep. In fact, the EEG changes in this type of sedation are similar to the EEG changes in natural sleep (5).
Cooperative sedation with dexmedetomidine is very different than the sedation produced by GABAergic drugs (benzodiazepines and propofol), where arousal occurs only after the drug is discontinued and the sedative effects abate. In fact, daily interruption of benzodiazepine sedation is aimed at achieving the type of sedation produced by dexmedetomidine; i.e., arousable and cooperative. Dexmedetomidine should be well suited for ventilator-dependent patients, because sedation can be continued during the transition period from mechanical ventilation to spontaneous breathing.
Delirium
Clinical studies have shown a lower prevalence of delirium in patients who are sedated with dexmedetomidine instead of midazolam (55), and based on these studies, dexmedetomidine is recommended over benzodiazepines for the sedation of patients with ICU-acquired delirium (3).
Adverse Effects
Dexmedetomidine produces dose-dependent decreases in heart rate, blood pressure, and circulating norepinephrine levels (sympatholytic effect) (5). Patients with heart failure and cardiac conduction defects are particularly susceptible to the sympatholytic actions of dexmedetomidine. Life-threatening bradycardia has been reported, primarily in patients treated with high infusion rates of dexmedetomidine (>0.7 µg/kg/min) together with a loading dose (56 ). Patients with cardiac conduction defects should not receive dexmedetomidine, and patients with heart failure or hemodynamic instability should not receive a loading dose of the drug.
Haloperidol
Haloperidol (Haldol) is a first-generation antipsychotic agent that has a long history of treating agitation and delirium (57–58).
Features
Haloperidol produces its sedative and antipsychotic effects by blocking dopamine receptors in the central nervous system. Following an intravenous bolus dose of haloperidol, sedation is evident in 10–20 minutes, and the effect lasts 3–4 hours. There is no respiratory depression, and hypotension is unusual in the absence of hypovolemia.
DOSING: The recommended dosing for intravenous haloperidol is shown in Table 51.7 (59,60). Because haloperidol has a delayed onset of action, midazolam can be given with the first dose of haloperidol to achieve more rapid sedation. Individual patients show a wide variation in serum drug levels after a given dose of haloperidol (61). If there is no evidence of a sedative response after 10 minutes, the dose should be doubled. If there is a partial response at 10–20 minutes, a second dose can be given along with 1 mg lorazepam (preferred to midazolam because of the longer duration of action) (61). Lack of response to a second dose of haloperidol should prompt a switch to another agent.
Table 51.7 Intravenous Haloperidol for the Agitated Patient

Adverse Effects
The adverse effects of haloperidol include: (a) extrapyramidal reactions, (b) neuroleptic malignant syndrome, and (c) ventricular tachycardia.
1. Extrapyramidal reactions (e.g., rigidity, spasmodic movements) are dose-related side effects of oral haloperidol therapy, but these reactions are uncommon when haloperidol is given intravenously (for unclear reasons) (61).
2. The neuroleptic malignant syndrome (described on pages 763–765) is an idiosyncratic reaction to neuroleptic agents that consists of hyperpyrexia, severe muscle rigidity, and rhabdomyolysis. This condition has been reported with intravenous haloperidol (62), but is rare.
3. The most publicized risk of therapy with haloperidol is prolongation of the QT interval on the electrocardiogram, which can trigger a polymorphic ventricular tachycardia (torsade de pointes, shown in Figure 15.8). This arrhythmia is reported in up to 3.5% of patients receiving intravenous haloperidol (63), which provides a reason to avoid haloperidol in patients with a prolonged QT interval.
A FINAL WORD
Improving the ICU Experience
It is not possible to completely eliminate the stress experienced by ICU patients because being critically ill is inherently stressful. However, the following considerations might help to reduce the “unpleasantness” of the ICU experience.
1. Critically ill patients experience pain in situations that are not normally painful. For example, being turned in bed is one of the most painful experiences reported by ICU patients, and up to 50% of patients experience pain at rest, without any noxious stimulus (8,10). This indicates that pain control is a full-time endeavor in ICU patients.
2. Unrelieved pain can be a source of agitation, so make sure that pain is relieved before considering a sedative drug for agitation.
3. When a benzodiazepine is used for prolonged (>48 hrs) sedation, attention to preventing drug accumulation and prolonged sedation can result in a shorter time in the ICU. The preventive measures include daily interruption of drug infusions, or maintaining light sedation using a reliable sedation scale to guide adjustments in drug dosage.
4. Consider using dexmedetomidine for sedation, because this drug allows the patient to be aroused while still sedated (e.g., to help with repositioning or speak with family members). When arousal is no longer needed, the patient will resume the prior level of sedation. This is more like sleep than a drug-induced stupor.
5. Finally, communicate with patients (e.g., tell them what you are going to do before doing it), and allow some “down time” for patients to sleep.
REFERENCES
Introduction
1. Loper KA, Butler S, Nessly M, Wild L. Paralysed with pain: the need for education. Pain 1989; 37:315–316.
2. Rotondi AJ, Chelluri L, Sirio C, et al. Patients’ recollections of stressful experiences while receiving prolonged mechanical ventilation in an intensive care unit. Crit Care Med 2002; 30:746–752.
Clinical Practice Guideline
3. Barr J, Fraser GL, Puntillo K, et al. Clinical practice guidelines for the management of pain. agitation, and delirium in adult patients in the intensive care unit. Crit Care Med 2013; 41:263–306.
Reviews
4. Devlin JW, Roberts RJ. Pharmacology of commonly used analgesics and sedatives in the ICU: benzodiazepines, propofol, and opioids. Crit Care Clin 2009; 25:431–449.
5. Panzer O, Moitra V, Sladen RN. Pharmacology of sedative-analgesic agents: dexmedetomidine, remifentanil, ketamine, volatile anesthetics, and the role of peripheral mu antagonists. Crit Care Clin 2009; 25:451–469.
The ICU Experience
6. Granja C, Lopes A, Moreira S, et al. Patients’ recollections of experiences in the intensive care unit may affect their quality of life. Crit Care 2005; 9:R96–R109.
7. Samuelson KA, Lundberg D, Fridlund B. Stressful experiences in relation to depth of sedation in mechanically ventilated patients. Nurs Crit Care 2007; 12:93–104.
8. Payen J-F, Chanques G, Mantz J, et al, for the DOLOREA Investigators. Current practices in sedation and analgesia for mechanically ventilated critically ill patients. Anesthesiology 2007; 106:687–695.
9. Schelling G, Stoll C, Haller M, et al. Health-related quality of life and posttraumatic stress disorder in survivors of the acute respiratory distress syndrome. Crit Care Med 1998; 26:651–659.
10. Chanques G, Sebbane M, Barbotte E, et al. A prospective study of pain at rest: incidence and characteristics of an unrecognized symptom in surgical and trauma versus medical intensive care unit patients. Anesthesiology 2007; 107:858–860.
11. Pain terms: A list with definitions and notes on usage, recommended by the IASP subcommittee on taxonomy. Pain 1979; 6:249.
12. Ahlers S, van Gulik L, van der Veen A, et al. Comparison of different pain scoring systems in critically ill patients in a general ICU. Crit Care 2008; 12:R15. (An open source journal.)
13. Siffleet J, Young J, Nikoletti S, et al. Patients’ self-report of procedural pain in the intensive care unit. J Clin Nurs 2007; 16:2142–2148.
Pain Control with Opioids
14. Pasternak GW. Pharmacological mechanisms of opioid analgesics. Clin Neuropharmacol 1993; 16:1–18.
15. Veselis RA, Reinsel RA, Feshchenko VA, et al. The comparative amnestic effects of midazolam, propofol, thiopental, and fentanyl at equisedative concentrations. Anesthesiology 1997; 87:749–764.
16. Rosow CE, Moss J, Philbin DM, et al. Histamine release during morphine and fentanyl anesthesia. Anesthesiology 1982; 56:93–96.
17. Smith MT. Neuroexcitatory effects of morphine and hydromorphone: evidence implicating the 3-glucuronide metabolites. Clin Exp Pharmacol Physiol 2000; 27:524–528.
18. Aronoff GR, Berns JS, Brier ME, et al. Drug Prescribing in Renal Failure: Dosing Guidelines for Adults. 4th ed. Philadelphia: American College of Physicians, 1999.
19. Eschenbacher WL, Bethel RA, Boushey HA, Sheppard D. Morphine sulfate inhibits bronchoconstriction in subjects with mild asthma whose responses are inhibited by atropine. Am Rev Resp Dis 1984; 130:363–367.
20. Felden L, Walter C, Harder S, et al. Comparative clinical effects of hydromorphone and morphine: a meta-analysis. Br J Anesth 2011; 107:319–328.
21. Latta KS, Ginsberg B, Barkin RL. Meperidine: a critical review. Am J Therap 2002; 9:53–68.
22. White PF. Use of patient-controlled analgesia for management of acute pain. JAMA 1988; 259:243–247.
23. Bowdle TA. Adverse effects of opioid agonists and agonist-antagonists in anaesthesia. Drug Safety 1998; 19:173–189.
24. Weil JV, McCullough RE, Kline JS, et al. Diminished ventilatory response to hypoxia and hypercapnia after morphine in normal man. N Engl J Med 1975; 292:1103–1106.
25. Bailey PL. The use of opioids in anesthesia is not especially associated with nor predictive of postoperative hypoxemia. Anesthesiology 1992; 77:1235.
26. Schug SA, Zech D, Grond S. Adverse effects of systemic opioid analgesics. Drug Safety 1992; 7:200–213.
27. Tomicheck RC, Rosow CE, Philbin DM, et al. Diazepam-fentanyl interaction—hemodynamic and hormonal effects in coronary artery surgery. Anesth Analg 1983; 62:881–884.
28. Meissner W, Dohrn B, Reinhart K. Enteral naloxone reduces gastric tube reflux and frequency of pneumonia in critical care patients during opioid alagesia. Crit Care Med 2003; 31:776–780.
Non-Opioid Analgesics
29. Buckley MM, Brogden RN. Ketorolac. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential. Drugs 1990; 39:86–109.
30. Gillis JC, Brogden RN. Ketorolac. A reappraisal of its pharmacodynamic and pharmacokinetic properties and therapeutic use in pain management. Drugs 1997; 53:139–188.
31. Ketorolac Tromethamine. In: McEvoy GK, ed. AHFS Drug Information, 2012. Bethesda: American Society of Health System Pharmacists, 2012:2139–2148.
32. Ready LB, Brown CR, Stahlgren LH, et al. Evaluation of intravenous ketorolac administered by bolus or infusion for treatment of postoperative pain. A double-blind, placebo-controlled, multicenter study. Anesthesiology 1994; 80:1277–1286.
33. Strom BL, Berlin JA, Kinman JL, et al. Parenteral ketorolac and risk of gastrointestinal and operative site bleeding. A postmarketing surveillance study. JAMA 1996; 275:376–382.
34. Reinhart DI. Minimising the adverse effects of ketorolac. Drug Safety 2000; 22:487–497.
35. Scott LJ. Intravenous ibuprofen. Drugs 2012; 72:1099–1109.
36. Yeh YC, Reddy P. Clinical and economic evidence for intravenous acetaminophen. Pharmacother 2012; 32:559–579.
37. Sinatra RS, Jahr JS, Reynolds LW, et al. Efficacy and safety of single and repeated administration of 1 gram intravenous acetaminophen injection (paracetamol) for pain management after major orthopedic surgery. Anesthesiology 2005; 102:822–831.
Anxiety in the ICU
38. Ely EW, Inouye SK, Bernard GR, et al. Delirium in mechanically ventilated patients: validity and reliability of the confusion assessment method for the intensive care unit (CAM-ICU). JAMA 2001;286:2703–2710.
39. Sessler CN Gosnell MS, Grap MJ, et al. The Richmond Agitation-Sedation Scale: validity and reliability in adult intensive care units. Am J Resp Crit Care Med 2002; 166:1338–1344.
40. Ely EW, Truman B, Shintani A, et al. Monitoring sedation status over time in ICU patients: reliability and validity of the Richmond Agitation-Sedation Scale (RASS). JAMA 2003; 289:2983–2991.
Benzodiazepines
41. Reves JG, Fragen RJ, Vinik HR, et al. Midazolam: pharmacology and uses. Anesthesiology 1985; 62:310–324.
42. Pohlman AS, Simpson KP, Hall JB. Continuous intravenous infusions of lorazepam versus midazolam for sedation during mechanical ventilatory support: a prospective, randomized study. Crit Care Med 1994; 22:1241–1247.
43. Kress JP, Pohlman AS, O’Connor MF, et al. Daily interruption of sedative infusions in critically ill patients undergoing mechanical ventilation. N Engl J Med 2000; 342:1471–1477.
44. Zaal IJ, Slooter AJC. Delirium in critically ill patients: epidemiology, pathophysiology, diagnosis and management. Drugs, 2012; 72:1457–1471.
45. Wilson KC, Reardon C, Theodore AC, Farber HW. Propylene glycol toxicity: a severe iatrogenic illness in ICU patients receiving IV benzodiazepines. Chest 2005; 128:1674–1681.
46. Arroglia A, Shehab N, McCarthy K, Gonzales JP. Relationship of continuous infusion lorazepam to serum propylene glycol concentration in critically ill adults. Crit Care Med 2004; 32:1709–1714.
47. Nordt SP, Vivero LE. Pharmaceutical additives. In: Nelson LS, Lewin NA, Howland MA, et al, eds. Goldfrank’s Toxicological Emergencies. 9th ed, New York:McGraw Hill, 2011:803–816.
48. Shafer A. Complications of sedation with midazolam in the intensive care unit and a comparison with other sedative regimens. Crit Care Med 1998; 26:947–956.
Propofol
49. McKeage K, Perry CM. Propofol: a review of its use in intensive care sedation of adults. CNS drugs 2003; 17:235–272.
50. Riker RR, Fraser GL. Adverse events associated with sedatives, analgesics, and other drugs that provide patient comfort in the intensive care unit. Pharmacotherapy 2005; 25:8S–18S.
51. Devaud JC, Berger MM, Pannatier A. Hypertriglyceridemia: a potential side effect of propofol sedation in critical illness. Intensive Care Med 2012; 38:1990–1998.
52. Fong JT, Sylvia L, Ruthazer R, et al. Predictors of mortality in patients with suspected propofol infusion syndrome. Crit Care Med 2008; 36:2281–2287.
53. Fodale V, LaMonaca E. Propofol infusion syndrome: an overview of a perplexing disease. Drug Saf 2008; 31:293–303.
Dexmedetomidine
54. Bhana N, Goa KL, McClellan KJ. Dexmedetomidine. Drugs 2000; 59:263–268.
55. Riker RR, Shehabi Y, Bokesch PM, et al. SEDCOM (Safety and Efficacy of Dexmedetomidine Compared With Midazolam) Study Group: Dexmedetomi-dine vs. midazolam for sedation of critically ill patients. JAMA 2009; 301:489–499.
56. Tan JA, Ho KM. Use of dexmedetomidine as a sedative and analgesic agent in critically ill patients: a meta-analysis. Intensive Care Med 2010; 36:926–939.
Haloperidol
57. Haloperidol. In: McEvoy GK, ed. AHFS Drug Information, 2012. Bethesda: American Society of Health System Pharmacists, 2012:2542–2547.
58. Clinton JE, Sterner S,Steimachers Z, Ruiz E. Haloperidol for sedation of disruptive emergency patients. Ann Emerg Med 1987; 16:319–322.
59. Jacobi J, Fraser GL, Coursin DB, et al. Clinical practice guidelines for the sustained use of sedatives and analgesics in the critically ill adult. Crit Care Med 2002; 30:119–141.
60. Riker RR, Fraser GL, Cox PM. Continuous infusion of haloperidol controls agitation in critically ill patients. Crit Care Med 1994; 22:433–440.
61. Sanders KM, Minnema AM, Murray GB. Low incidence of extrapyramidal symptoms in the treatment of delirium with intravenous haloperidol and lorazepam in the intensive care unit. J Intensive Care Med 1989; 4:201–204.
62. Sing RF, Branas CC, Marino PL. Neuroleptic malignant syndrome in the intensive care unit. J Am Osteopath Assoc 1993; 93:615–618.
63. Sharma ND, Rosman HS, Padhi ID, et al. Torsade de Pointes associated with intravenous haloperidol in critically ill patients. Am J Cardiol 1998; 81:238–240.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


