Diagnosis
The diagnosis of acute pancreatitis requires: (a) an increase in serum levels of pancreatic enzymes (amylase and lipase) to at least 3 times the upper limit of normal, and (b) evidence of pancreatitis on contrast-enhanced computed tomography (1).
Pancreatic Enzymes
AMYLASE: Amylase is an enzyme that cleaves starch into smaller polysaccharides. The principal sources of amylase are the pancreas, salivary glands, and fallopian tubes. Serum amylase levels begin to rise 6–12 hours after the onset of acute pancreatitis, and they return to normal in 3–5 days. An increase in serum amylase levels to 3 times the upper limit of normal (the threshold for the diagnosis of acute pancreatitis) has a high sensitivity (>90%) but a low specificity (as low as 70%) for the diagnosis of acute pancreatitis (6).
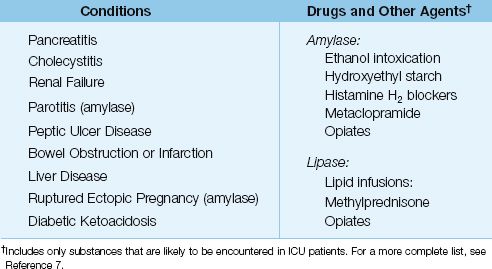
The low specificity of serum amylase is a reflection of the numerous conditions that can elevate serum amylase levels. These are listed in Table 39.2 (7). About 25% of the nonpancreatic conditions in this table can result in serum amylase levels that overlap those seen in acute pancreatitis (8). The ones that deserve mention include parotitis, ruptured ectopic pregnancy, and acute alcohol intoxication. Of particular note, hyperamylasemia of salivary origin is reported in 40% of cases of acute alcohol intoxication (6). (Note: The reference range for serum amylase is not mentioned because it often varies in different clinical laboratories.)
Table 39.2 Sources of Elevated Serum Amylase and Lipase Levels
LIPASE: Lipase is an enzyme that hydrolyses triglycerides to form glycerol and free fatty acids. The principal sources of lipase are the tongue, pancreas, liver, intestine, and circulating lipoproteins. In acute pancreatitis, serum lipase levels begin to rise earlier than serum amylase (at 4 to 8 hours), and the serum levels remain elevated longer than serum amylase (for 8 to 14 days).
Like amylase, there are several nonpancreatic conditions that can elevate serum lipase levels, as shown in Table 39.2. However, unlike amylase, nonpancreatic conditions rarely raise serum lipase levels high enough to overlap with the levels seen in acute pancreatitis (8). Therefore, serum lipase is considered more specific than serum amylase for the diagnosis of acute pancreatitis. An increase in serum lipase to three times the upper limit of normal has a sensitivity and specificity of 80–100% for acute pancreatitis (6).
RECOMMENDATION: The serum lipase can be used alone for the diagnostic evaluation of pancreatitis. Adding the serum amylase assay does not increase diagnostic accuracy (6). However, pancreatic enzyme assays cannot be used to evaluate the severity of illness (6).
Computed Tomography
Contrast-enhanced computed tomography (CT) is the most reliable diagnostic test for acute pancreatitis, and can identify the type of pancreatitis (edematous vs. necrotizing) as well as localized complications (e.g., infection). Figure 39.1 shows a contrast-enhanced CT image of edematous pancreatitis. The pancreas is thickened and enhances completely, and the border of the pancreas is blurred, which is characteristic of pancreatic edema. Compare this with the image in Figure 39.2, which shows a large area that is not contrast-enhanced in the region of the neck and body of the pancreas. This represents pancreatic necrosis, and identifies the condition as necrotizing pancreatitis. The full extent of pancreatic necrosis may not be evident on CT imaging for the first week after the onset of symptoms (1), so repeat imaging is advised in patients with persistent symptoms or severe pancreatitis.
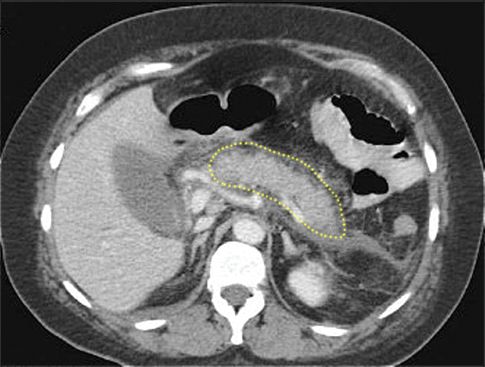
FIGURE 39.1 Contrast-enhanced CT image showing edematous pancreatitis. The pancreas (outlined by the dotted line) is enlarged and enhances completely. There is also blurring of the pancreatic border, which is characteristic of edema formation. Image digitally enhanced.
When IV contrast cannot be administered (because of a dye allergy or a serum creatinine above 1.5 mg/dL) CT imaging is less likely to distinguish between edematous and necrotizing pancreatitis.
Biliary Evaluation
Since gallstones are the leading cause of acute pancreatitis in the United States (4), an evaluation of the gall bladder and biliary tree is advised in all cases of confirmed acute pancreatitis. Contrast-enhanced CT images may suffice for this evaluation, but in cases where a CT scan is inconclusive, or has not been performed, ultrasonography is recommended.
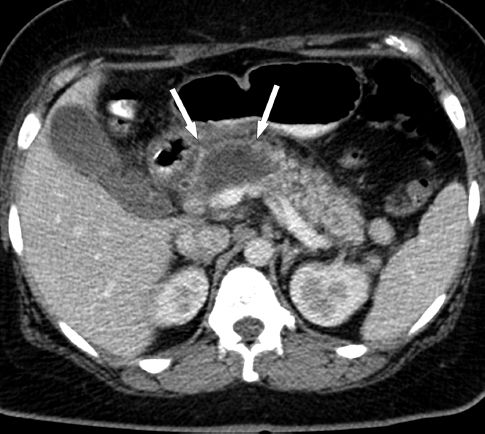
FIGURE 39.2 Contrast-enhanced CT image showing necrotizing pancreatitis. The area that is not contrast-enhanced (indicated by the arrows) represents necrosis in the neck and body of the pancreas. Image from Reference 1.
SEVERE PANCREATITIS
Severe pancreatitis is defined as acute (usually necrotizing) pancreatitis that is associated with persistent (>48 hrs) injury in at least one other organ system (1). The cause of the extrapancreatic organ injury is progressive systemic inflammation (similar to severe sepsis and septic shock), and the organs that are typically involved include the lungs (acute respiratory distress syndrome, or ARDS), the kidneys (acute kidney injury), and the circulatory system (hypotension and circulatory shock). Pancreatic enzymes and CT images have a poor correlation with the clinical severity of this condition
The management of severe pancreatitis is best conducted in an ICU setting, and includes the following measures: (a) removing the precipitating condition (e.g., obstructing gallstones), (b) providing supportive care for the extrapancreatic organ injuries (e.g., mechanical ventilation for ARDS), (c) early nutritional support with enteral tube feedings, and (d) managing intraabdominal complications (e.g., infection).
Circulatory Support
Circulatory support includes volume resuscitation and vasopressor drugs, if necessary.
Fluid Therapy
Severe pancreatitis is associated with loss of intravascular fluid through leaky systemic capillaries, and the resulting hypovolemia can produce additional pancreatic necrosis. For this reason, aggressive fluid therapy has been recommended early in the course of severe pancreatitis (9). There is no agreement on which type of fluid (colloid or crystalloid) is best, but crystalloid fluids are currently the popular choice. The initial regimen for volume resuscitation is summarized as follows:
1. For crystalloid fluids, infuse 20 mL/kg (about 1.5 liters) over 60 to 90 minutes.
2. Follow with an infusion rate up to 250 mL/hr for the next 24–48 hours, to maintain a mean arterial pressure ≥65 mm Hg, and a urine output ≥0.5 mL/kg/hr.
CAUTION: Aggressive volume infusion has not been shown to improve outcomes in severe pancreatitis (10), and this practice can be deleterious by promoting edema formation, which can aggravate conditions like ARDS, and increases the risk of abdominal compartment syndrome. Therefore, after the initial volume infusion of 20 mL/kg, the infusion rate should be titrated to the desired blood pressure and urine output, but should not exceed 250 mL/hr. If volume infusion does not achieve the desired hemodynamic goals, vasopressor therapy should be initiated.
Vasopressor Therapy
There are no official recommendations regarding vasopressor therapy in severe pancreatitis, but norepinephrine is an appropriate choice. The initial infusion rate is 0.1 µg/kg/hr, which is then titrated to maintain a mean arterial pressure ≥65 mm Hg. All vasoconstrictor drugs can reduce splanchnic blood flow (especially phenylephrine), and could aggravate pancreatic necrosis, so careful titration of infusion rates (and avoiding phenylephrine) is advised.
Prophylactic Antibiotics
About one-third of patients with necrotizing pancreatitis develop infections in the necrotic areas of the pancreas (11). The pathogens are almost always Gram-negative enteric organisms, and the infections typically appear 7–10 days after the onset of illness. These infections are difficult to eradicate, and they are associated with increased mortality rates (11). Unfortunately, antibiotic prophylaxis does not reduce the incidence of pancreatic infections, and does not influence the mortality rate in severe pancreatitis (12). As a result, prophylactic antibiotics are not recommended in necrotizing pancreatitis (11).
Nutrition Support
Nutrition support should be started early (within 48 hours after the onset of illness) using enteral tube feedings, if possible (13).
Enteral Nutrition
The preference for enteral nutrition is based on the ability of tube feedings to exert a trophic effect on the bowel mucosa. This helps to maintain the structural and functional integrity of the bowel mucosa, and thereby reduce the risk of bacterial translocation across the bowel wall (which is considered the major route leading to pancreatic infections). Clinical studies have shown that enteral nutrition is associated with fewer infections, less multiorgan failure, and a lower mortality rate than total parenteral nutrition in patients with severe pancreatitis (14). (The effect of enteral feedings on the bowel mucosa is described in more detail in Chapter 48.)
FEEDING SITE: Tube feedings should be infused into the jejunum using long feeding tubes that can be placed with fluoroscopic or endoscopic guidance. Alternately, a feeding jejunostomy can be created in patients who require laparotomy for pancreatic debridement. Nasogastric feedings are not currently recommended, although one small study has shown no apparent harm from nasogastric feedings in severe pancreatitis (15).
FEEDING REGIMEN: The jejunum does not have the reservoir capacity of the stomach, so jejunal feedings should be advanced more slowly than gastric feedings. The diluting effect of gastric secretions is also lost in the jejunum, so isotonic feeding formulas are preferred to hypertonic formulas. Standard (polymeric) tube feedings can be used for jejunal feedings (13), but in patients with diarrhea, elemental feeding formulas may be preferred. (Elemental formulas are low in fat, and the protein is available as individual amino acids, which are presumably easier to digest.)
Abdominal Complications
Pancreatic Infection
The appearance of infection in necrotizing pancreatitis is often heralded by reappearance, persistence, or progression of systemic inflammation and multiorgan failure at 7–10 days after the onset of illness. A contrast-enhanced CT scan may show gas bubbles in the necrotic areas of the pancreas, as shown in Figure 39.3. If infection is suspected but gas bubbles are not evident on CT imaging, cultures must be obtained from the necrotic areas of the pancreas (using CT-guided needle aspiration). The treatment of choice for infected pancreatic necrosis is surgical debridement (necrosectomy) (11).
Abdominal Compartment Syndrome
There are several sources of increased intraabdominal pressure in severe pancreatitis, including peripancreatic fluid collections, ascites, and edema of the bowel wall (which is exaggerated by aggressive volume infusion). Abdominal compartment syndrome (ACS) has been reported in as many as 55% of patients with severe pancreatitis (16), but this may be an exaggeration because it is based on studies that used a relatively low abdominal pressures (15–20 mm Hg) for the diagnosis of ACS. Nevertheless, ACS is more common than suspected in severe pancreatitis, and measurements of abdominal pressure are warranted in any patient with acute pancreatitis who develops acute oliguric renal failure. (See Chapter 34 for more information on ACS.)
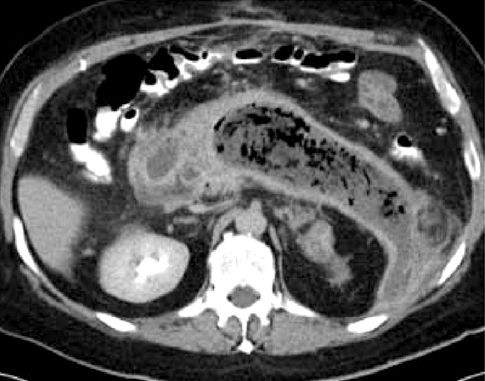
FIGURE 39.3 Contrast-enhanced CT image showing extensive necrosis of the pancreas with numerous gas bubbles, indicating infection.
Gallstone Pancreatitis
When acute pancreatitis is associated with gallstones, early endoscopic retrograde cholangio-pancreatography (ERCP) is indicated for biliary obstruction or evidence of cholangitis (i.e., fever and increasing liver enzymes) (17).
LIVER FAILURE
Types of Liver Failure
There are two types of liver failure that can appear in the ICU: (a) acute liver failure, and (b) acute-on-chronic liver failure.
Acute Liver Failure
Acute liver failure is an abrupt and rapid deterioration in liver function that occurs de novo, without prior liver disease. This is an uncommon condition with an annual incidence of 1 to 6 cases per million people in developed countries (18). Most cases are the result of viral hepatitis or drug-induced liver injury, and the principal clinical manifestation is severe hepatic encephalopathy. The leading cause of acute liver failure in the United States is acetaminophen overdose. (See Chapter 54 for a description of acetaminophen hepatotoxicity.)
Acute-on-Chronic Liver Failure
Most cases of liver failure involve patients with chronic liver disease (cirrhosis) who develop an abrupt deterioration in liver function as a result of a precipitating condition, usually an infection or variceal hemorrhage (19). The clinical manifestations often include signs of systemic inflammation (fever, leukocytosis, etc.), worsening ascites, mental status changes (hepatic encephalopathy), and a deterioration in renal function. The evaluation and management of this group of patients is described in the following text. The mortality rate in these patients is considerable, and ranges from 35% to 70% (19,20).
Spontaneous Bacterial Peritonitis
In patients with acute-on-chronic liver failure and ascites, 10% to 27% have evidence of infection in the ascitic fluid without an apparent primary site of infection (21). This condition is called spontaneous bacterial peritonitis (SBP), and the presumed mechanism is translocation of enteric pathogens across the bowel mucosa and into the peritoneal fluid. Cirrhosis predisposes to SBP because it impairs the normal function of the liver in eradicating enteric pathogens that translocate across the bowel wall. A single organism is isolated in most cases of SBP, and the isolates are Gram-negative aerobic bacilli (especially Escherichia coli) in 75% of cases, and Gram-positive aerobic cocci (especially streptococcal species) in 25% of cases (21).
Clinical Features
Fever, abdominal pain, and rebound tenderness are present in at least 50% of cases of SBP, but the condition can be asymptomatic in one-third of cases (21).
Diagnostic Approach
A diagnostic paracentesis should be performed on all patients with cirrhosis and ascites who are admitted for acute-on-chronic liver failure. An absolute neutrophil count ≥250 cells/mm3 in the ascitic fluid is presumptive evidence of infection, and is an indication to begin empiric antimicrobial therapy. Culture samples of ascitic fluid should be injected directly into blood culture bottles at the bedside because standard culture methods have a diagnostic yield of only 50% in cases of probable SBP (22).
Management
The preferred antibiotic for suspected SBP is cefotaxime (2 grams IV every 8 hours), or another third-generation cephalosporin (21–23). Unfortunately, the mortality rate in SBP is 30 to 40% despite adequate antibiotic coverage (22); this may be explained by the fact that 30% of patients with SBP develop hepatorenal syndrome (23), which has a mortality rate in excess of 50% (see later for a description of this syndrome).
ALBUMIN INFUSIONS: Because renal hypoperfusion plays an important role in the pathogenesis of hepatorenal syndrome (see later), clinical studies have evaluated the role of albumin infusions in SBP. The results of these studies indicate that infusions of albumin can reduce the incidence of hepatorenal syndrome in SBP, but only in high-risk patients; i.e., those with a BUN >30 mg/dL, creatinine >1 mg/dL and bilirubin >4 mg/dL (24). The recommended albumin infusion regimen is as follows (24):
| Day 1: | 1.5 g /kg body weight, infused within 6 hours of the diagnosis of SBP. |
| Day 3: | 1.0 g/kg body weight. |
It is unclear at this time if the benefit from albumin is a volume effect, or is related to some other effect (e.g., albumin has antioxidant activity, and can also bind cytokines).
Management of Ascites
The formation of ascites in patients with cirrhosis is partly the result of sodium retention by the kidneys in response to activation of the renin-angiotensin-aldosterone system. The management of ascites is aimed at counteracting this sodium retention using diuretics (furosemide and spironolactone) and restricted sodium intake.
Sodium Restriction
Daily intake of sodium should be restricted to 2 grams (88 mEq), if possible (22). This is often an unrealistic goal in hospitalized patients (who require saline or Ringer’s lactate infusions for a variety of reasons), but higher sodium intakes can be tolerated when urinary sodium losses exceed 88 mEq daily (i.e., during diuretic therapy). Fluid restriction is not necessary unless it is needed for symptomatic hyponatremia.
Spironolactone
The actions of aldosterone to promote sodium retention in cirrhotic patients can be antagonized by spironolactone. The drug is given orally (or via feeding tube) at an initial dose of 100 mg once daily. The daily dose can be increased in 100 mg increments, if needed, to a maximum daily dose of 400 mg. The use of spironolactone alone is not advised because of the risk of hyperkalemia (22).
Furosemide
Diuretic therapy with furosemide promotes urinary sodium loss, and also reduces the risk of hyperkalemia from spironolactone. The initial dose is 40 mg (oral or intravenous) daily, and this can be increased gradually, in 40 mg increments, to a maximum daily dose of 160 mg, if necessary. Furosemide should not be used alone because it is less effective than spironolactone for treating cirrhosis-related ascites (22).
Large-Volume Paracentesis
Patients with tense ascites can receive immediate relief from large-volume paracentesis. A volume of 5 liters can be removed at one “sitting” without adverse hemodynamic consequences (25). If larger volumes are removed, albumin can be infused at a dose of 8.5 mg/kg for each liter of fluid removed (23).
End-Points
There is no ceiling for daily weight loss in cirrhotic patients with edema and ascites (22). Fluid loss is permitted until the baseline or premorbid body weight is achieved, or until there is evidence of prerenal azotemia. An increase in serum creatinine to 2 mg/dL is an indication to discontinue diuretic therapy. Avoiding excessive diuresis is an important consideration for limiting the risk of hepatorenal syndrome (see later).
About 10% of patients with cirrhosis and ascites are resistant to diuretic therapy (22). The prognosis in this situation is poor, and liver transplantation should be considered.
Hepatorenal Syndrome
Hepatorenal syndrome (HRS) is a functional renal failure (i.e., occurs without intrinsic renal disease) that occurs in patients with advanced cirrhosis, especially those with spontaneous bacterial peritonitis or sepsis from another source (26).
Pathogenesis
HRS is the result of hemodynamic alterations in the splanchnic and renal circulations. Cirrhosis is associated with splanchnic vasodilation, and the neurohumoral (renin system) response to this vasodilation results in vasoconstriction in other organs, including the kidneys (26). The renal vasoconstriction creates a situation where the glomerular filtration rate is vulnerable to small decrements in cardiac output. Sepsis is also associated with splanchnic vasodilation, which could explain the association between sepsis and HRS.
Diagnosis
The diagnostic criteria for HRS are shown in Table 39.3. The criteria include renal impairment (serum creatinine >1.5 mg/dL) that does not respond to albumin infusions, and no other likely source of renal dysfunction (i.e., nephrotoxic drugs, shock, or parenchymal kidney disease).
Table 39.3 Clinical Approach to Hepatorenal Syndrome
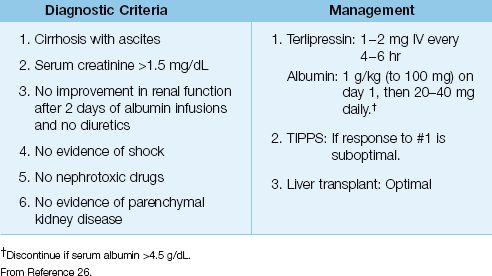
Management
The management of HRS is designed to correct the hemodynamic changes that are responsible for HRS. First-line therapy includes a splanchnic vasoconstrictor (terlipressin, a vasopressin analogue) and a volume expander (albumin). An effective dosing regimen is shown in Table 39.3. Over 50% of patients with HRS will show improvement in renal function with this regimen (26,27). However, HRS often relapses after drug therapy is discontinued, and long-term survival requires liver transplantation (26). Transjugular intrahepatic portosystemic stent-shunt (TIPPS) can improve renal function in HRS (26), but this procedure promotes hepatic encephalopathy, so it is reserved for transplant candidates that are unresponsive to pharmacotherapy.
HEPATIC ENCEPHALOPATHY
Liver failure produces an encephalopathy that is characterized by cerebral edema, disordered thinking, and altered consciousness. Hepatic encephalopathy is the dominant manifestation in acute liver failure, whereas in acute-on-chronic liver failure, the encephalopathy is usually preceded by an acute insult (e.g., variceal hemorrhage). Ammonia has been identified as a key factor in the pathogenesis of hepatic enceph-alopathy (28).
Pathogenesis
Ammonia (NH3) is a byproduct of protein degradation, and is produced primarily in the bowel (and to a lesser degree in skeletal muscle and kidneys). The liver plays a major role in clearing ammonia by converting it to urea in the urea cycle. This clearance mechanism is impaired or lost in liver failure, resulting in a progressive rise in ammonia levels in blood. Am-monia eventually crosses the blood-brain barrier and is taken up by astrocytes, which use the ammonia to convert glutamate to glutamine; i.e.,
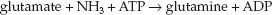
(Astrocytes normally provide glutamine for neurons, which use the glutamine as a substrate for the production of glutamate, a neurotransmitter.) The ammonia load in astrocytes leads to accumulation of glutamine, and this creates an osmotic force that draws water into the astrocytes. The result is cerebral edema, astrocyte damage, and impaired synaptic transmission in the brain.
Clinical Features
The principal features of progressive hepatic encephalopathy are shown in Table 39.4 (29). Agitation and disorientation are prominent in the early stages, while depressed consciousness is the dominant feature in the late stages. The cranial nerves are not affected, but dysarthria can be present (30). Involuntary movements like tremors or asterixis (clonic movements during wrist extension) can appear, while sensation is intact. Focal neurological deficits are considered evidence of an alternative diagnosis (30).
Table 39.4 Progressive Stages of Hepatic Encephalopathy
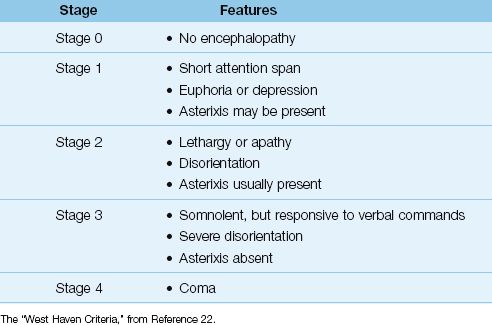
Diagnostic Evaluation
The diagnosis of hepatic encephalopathy is usually made by excluding other causes of altered mentation. Other conditions that should be considered in patients with cirrhotic liver failure include drug overdose, subdural hematoma, and Wernicke’s encephalopathy (from thiamine deficiency). Neuroimaging studies are performed to eliminate other diagnoses. The only diagnostic test that can help to identify hepatic enceph-alopathy is the serum ammonia level.
Serum Ammonia
Considering the role played by ammonia in the pathogenesis of hepatic encephalopathy, it is no surprise that serum ammonia levels are typically elevated in patients with hepatic encephalopathy. This is demonstrated in Figure 39.4, which shows the relationship between ammonia levels in blood (arterial and venous) and the presence and severity of hepatic encephalopathy (31). Although the ammonia levels are mildly elevated in the absence of hepatic encephalopathy (stage 0), the levels are higher in the presence of hepatic encephalopathy, and the degree of elevation corresponds to the severity of the condition. Note that the ammonia levels are higher in arterial blood. Although the difference between arterial and venous ammonia levels was not statistically significant in this study (31), arterial blood seems optimal for identifying hepatic encephalopathy in the early stages of the condition.
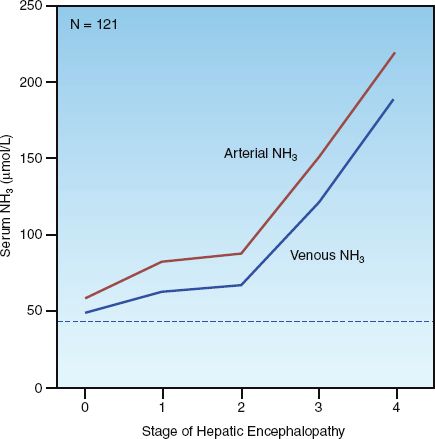
FIGURE 39.4 Relationship between arterial and venous ammonia (NH3) and the presence and severity of hepatic encephalopathy. The stages of encephalopathy correspond to those in Table 39.3. The horizontal dotted line represents the upper limit of the normal range for serum ammonia (47 µmol/L) at the study hospital. N indicates number of patients studied. Data from Reference 31.
Treatment
The treatment of hepatic encephalopathy is aimed at reducing the ammonia burden in the central nervous system. The most effective strategy is to impair ammonia production in the bowel with lactulose (first-line therapy) and nonabsorbable antibiotics (second-line therapy).
Lactulose
Lactulose is a nonabsorbable disaccharide that is metabolized by “lactic acid bacteria” (e.g., Lactobacillus acidophilus) in the bowel (32). This promotes the formation of short-chain fatty acids, and the resulting acidification of the bowel lumen reduces the ammonia burden from the bowel in two ways: (a) by eradicating ammoniagenic microorganisms (mostly Gram-negative aerobic bacilli), and (b) by reducing ammonia absorption from the bowel. (The bactericidal actions of an acid pH are illustrated in Figure 5.3.) The dosing recommendations for lactulose in acute hepatic encephalopathy are as follows (29):
1. Oral or Nasogastric Route: Start with 45 mL lactulose every hour until evacuation occurs, then reduce dose to 30 mL every 8–12 hours. This is the preferred route.
2. Retention Enema: Mix 300 mL lactulose in one liter of tap water. Administer by high rectal enema, and retain for one hour with the patient in the Trendelenburg position.
Lactulose can promote an osmotic diarrhea, and the dosage should be reduced (or temporarily halted) if diarrhea appears. In patients with diarrhea at the outset, lower doses of lactulose can be combined with a nonabsorbable antibiotic.
Nonabsorbable Antibiotics:
Nonabsorbable antibiotics are used to eradicate ammonia-generating organisms (i.e., Gram-negative aerobic bacilli). The following are 2 regimens that can be used in acute hepatic encephalopathy:
1. Neomycin: The oral (nasogastric) dose is 3 to 6 grams daily in 3 divided doses, and continued for 1 to 2 weeks (23).
2. Rifamaxin: A regimen with proven success is 1,200 mg daily (400 mg by mouth or nasogastric tube every 8 hours) for 10–21 days (33).
Neomycin is the traditional choice (and is devoid of oto- and nephrotoxic effects when used in short-term therapy), while rifamaxin (a rifampin analogue with broad-spectrum activity and little toxicity) is rapidly gaining in popularity. There is currently no evidence of superiority with either drug regimen.
Nutrition Support
Protein restriction (which would reduce the ammonia burden from the bowel) is not recommended for patients with hepatic encephalopathy because these patients have increased rates of protein catabolism, and restricting protein intake would promote a negative nitrogen balance (34). The recommended protein intake in critically ill patients is 1.2 to 1.5 g/kg/day (see Chapter 47), so staying at the low end of this range (1.2 g/kg/day) might be the best choice in patients with hepatic encephalopathy.
A FINAL WORD
Back to the Bowel
One of the recurring themes in this book is the importance of the bowel as a source of infection in critically ill patients (see Chapters 5 and 40). Two observations in this chapter demonstrate the normal defense mechanisms that prevent infections of bowel origin.
The first observation is the ability of enteral tube feedings to reduce the incidence of sepsis and multiorgan failure in patients with severe pancreatitis. This highlights the trophic effect of bulk nutrients on the structural and functional integrity of the mucosal barrier in the gut; i.e., the “non-nutritive” function of enteral feeding. (See Chapter 48 for more information on this topic.)
The second observation is the occurrence of spontaneous bacterial peritonitis in patients with cirrhosis and ascites. This is a classic example of an infection that is caused by the translocation of enteric pathogens across the bowel mucosa, and it highlights the importance of the reticuloendothelial system in the bowel (mostly represented by the liver) in defending against the spread of enteric pathogens.
REFERENCES
Pancreatitis
1. Banks PA, Bollen TL, Dervenis C, et al. Classification of acute pancreatitis – 2012: revision of the Atlanta classification and definitions by international consensus. Gut 2012; 62:102–111.
2. Cavallini G, Frulloni L, Bassi C, et al. Prospective multicentre survey on acute pancreatitis in Italy (Proinf-AISP). Dig Liver Dis 2004; 36:205–211.
3. Greer SE, Burchard KW. Acute pancreatitis and critical illness. A pancreatic tale of hypoperfusion and inflammation. Crit Care Med 2009; 136:1413–1419.
4. Forsmark CE, Baille J. AGA Institute technical review on acute pancreatitis. Gastroenterol 2007; 132:2022–2044.
5. Yang AL, Vadhavkar S, Singh G, Omary MB. Epidemiology of alcohol-related liver and pancreatic disease in the United States. Arch Intern Med 2008; 168:649–656.
6. Yadav D, Agarwal N, Pitchumoni CS. A critical evaluation of laboratory tests in acute pancreatitis. Am J Gastroenterol 2002; 97:1309–1318.
7. Gelrud D, Gress FG. Elevated serum amylase and lipase. UpToDate (accessed on May 30, 2013).
8. Gumaste VV, Roditis N, Mehta D, Dave PB. Serum lipase levels in nonpancreatic abdominal pain versus acute pancreatitis. Am J Gastroenterol 1993; 88:2051–2055.
Severe Pancreatitis
9. Tenner S. Initial management of acute pancreatitis: critical issues in the first 72 hours. Am J Gastroenterol 2004; 99:2489–2494.
10. Haydock MD, Mittal A, Wilms HR, et al. Fluid therapy in acute pancreatitis: anybody’s guess. Ann Surg 2013; 257:182–188.
11. Banks PA, Freeman ML, Practice Parameters Committee of the American College of Gastroenterology. Practice guidelines in acute pancreatitis. Am J Gastroenterol 2006; 101:2379–2400.
12. Hart PA, Bechtold ML, Marshall JB, et al. Prophylactic antibiotics in necrotizing pancreatitis: a meta-analysis. South Med J 2008; 101:1126–1131.
13. Parrish CR, Krenitsky J, McClave SA. Pancreatitis. 2012 A.S.P.E.N. Nutrition Support Core Curriculum. Silver Spring, MD: American Society of Parenteral and Enteral Nutrition, 2012:472–490.
14. Al-Omran M, AlBalawi ZH, Tashkandi MF, Al-Ansary LA. Enteral versus parenteral nutrition for acute pancreatitis. Cochrane Database Syst Rev 2010:CD002837.
15. Eatock FC, Chong P, Menezes N, et al. A randomized study of early nasogastric versus nasojejunal feeding in severe acute pancreatitis. Am J Gastro-enterol 2005; 100:432–439.
16. Al-Bahrani AZ, Abid GH, Holt A. et al. Clinical relevance of intra-abdominal hypertension in patients with severe acute pancreatitis. Pancreas 2008; 36:39–43.
17. Nathens AB, Curtis JR, Beale RJ, et al. Management of the critically ill patient with severe acute pancreatitis. Crit Care Med 2004; 32:2524–2536.
Liver Failure
18. Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet 2010; 376:190–201.
19. Olson JC, Kamath PS. Acute-on-chronic liver failure: concept, natural history, and prognosis. Curr Opin Crit Care 2011; 17:165–169.
20. Saliba F, Ichai P, Levesque E, Samuel D. Cirrhotic patients in the ICU: prognostic markers and outcome. Curr Opin Crit Care 2013; 19:154–160.
Ascites
21. Gilbert JA, Kamath PS. Spontaneous bacterial peritonitis: an update. Mayo Clin Proc 1995; 70:365–370.
22. Runyon BA. Management of adult patients with ascites caused by cirrhosis. Hepatology 1998; 27:264–272.
23. Moore CM, van Thiel DH. Cirrhotic ascites review: pathophysiology, diagnosis, and management. World J Hepatol 2013; 5:251–263.
24. Narula N, Tsoi K, Marshall JK. Should albumin be used in all patients with spontaneous bacterial peritonitis? Can J Gastroenterol 2011; 25:373–376.
25. Peltekian KM, Wong F, Liu PP, et al. Cardiovascular, renal, and neurohumoral responses to single large-volume paracentesis in cirrhotic patients with diuretic resistant ascites. Am J Gastroenterol 1997; 92:394–399.
Hepatorenal Syndrome
26. Dalerno F, Gerbes A, Gines P, et al. Diagnosis, prevention and treatment of hepatorenal syndrome in cirrhosis. Gut 2007l 56:131–1318.
27. Rajekar H, Chawla Y. Terlipressin in hepatorenal syndrome: evidence for present indications. J Gastroenterol Hepatol 2011; 26(Suppl):109–114.
Hepatic Encephalopathy
28. Clay AS, Hainline BE. Hyperammonemia in the ICU. Chest 2007; 132: 1368–1378.
29. Blei AT, Cordoba J, and the Practice Parameters Committee of the American College of Gastroenterology. Hepatic encephalopathy. Am J Gastroenterol 2001; 96:1968–1976.
30. Ferenci P, Lockwood A, Mullen K, et al. Hepatic encephalopathy – definition,nomenclature, diagnosis and quantification: Final report of the Working Party at the 11th World Congress of Gastroenterology, Vienna, 1998. Hepatol 2002; 55:716–721.
31. Ong JP, Aggarwal A, Krieger D, et al. Correlation between ammonia levels and the severity of hepatic encephalopathy. Am J Med 2003; 114:188–193.
32. Salminen S, Salminen E. Lactulose, lactic acid bacterial, intestinal microecology, and mucosal protection. Scand J Gastroenterol 1997; 222(Suppl):45–48.
33. Lawrence KR, Klee JA. Rifaximin for the treatment of hepatic encephalopathy. Pharmacotherapy 2008; 28:1019–1032.
34. Nutrition in end-stage liver disease: principles and practice. Gastroenterol-ogy 2008; 134:1729–1740.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


