The Carbon Age
2008
One of the rare sights in any ICU is a patient who is NOT receiving supplemental oxygen to breathe. Oxygen is used liberally in ICU patients, and oxygen therapy is guided by measures (i.e., the arterial PO2 and O2 saturation) that have no proven relationship with tissue oxygenation. As emphasized in this chapter, the excessive use of oxygen without evidence of impaired tissue oxygenation is problematic because it promotes the formation of toxic oxygen metabolites, which are capable of lethal cell injury.
This chapter begins with some insights on the design of the human body in relation to oxygen and oxygen therapy. This is followed by a description of the different oxygen delivery systems. The final section is devoted to the dark side of oxygen; i.e., the propensity for oxygen to damage aerobic organisms.
INSIGHTS
The Paucity of Oxygen in Tissues
Despite our dependence on oxygen for metabolic energy production, aerobic metabolism is carried out in an oxygen-restricted environment. Oxygen does not readily dissolve in water, which is why we need hemoglobin to transport oxygen, and this limits the volume of dissolved oxygen in the tissues of the body. The estimated volume of oxygen in the interstitial fluid and cells of the human body is shown in Table 22.1. According to these estimates, there is only about 13 mL of O2 in all the tissues of the human body!
Table 22.1 The Paucity of Dissolved Oxygen in Tissues

The estimates in Table 22.1 are based on the determinants of dissolved O2 in the equation shown below.
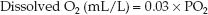 (22.1)
(22.1)
where 0.03 is the solubility coefficient of O2 in water (expressed as mL O2 per liter of body water per mm Hg of PO2) at a body temperature of 37°C. Experimental studies show that the intracellular PO2 is about 5 mm Hg (1) and interstitial PO2 is about 15 mm Hg (2). Using these PO2 values in equation 22.1, the concentration of dissolved O2 will be 0.03×5= 0.15 mL/L inside cells and 0.03×15=0.45 mL/L in the interstitial fluid. An average sized adult has an intracellular volume of about 23 liters and an interstitial fluid volume of about 16 liters, so the total volume of dissolved O2 will be 0.15×23=3.5 mL in cells and 0.45×16=9.6 mL in the interstitial fluid.
Man as a Microaerophilic Organism
Humans are described as obligate aerobic organisms; i.e., organisms that require oxygen to survive. However, according to the estimates in Table 22.1, humans are more accurately described as microaerophilic organisms; i.e., organisms that require only low concentrations of oxygen to survive.
Implications
The oxygen-restricted environment in tissues can be viewed as a safeguard against the damaging effects of oxygen metabolites (described later in the chapter). This safeguard will be threatened or lost if oxygen therapy exposes the tissues to more O2 than is needed to sustain aerobic metabolism. The remaining observations in this section are relevant to this issue.
Tolerance to Hypoxemia
Oxygen therapy is used to prevent hypoxemia (i.e., an arterial PO2 <60 mm Hg or an arterial O2 saturation <90%) (3). However, there is no evidence that hypoxemia impairs tissue oxygenation, regardless of severity (4–6). The data points in Figure 22.1 show the blood lactate levels in patients with severe hypoxemia (i.e., arterial PO2 <40 mm Hg) from an acute exacerbation of chronic obstructive lung disease (4). Note that the blood lactate levels fail to rise above 2 mmol/L (the threshold for hyperlactatemia) despite arterial PO2 levels as low as 22 mm Hg. This provides evidence that severe hypoxemia did not impair aerobic metabolism in these patients. Similar observations have been reported in patients with acute respiratory distress syndrome (5), indicating that tolerance to severe hypoxemia is not an adaptation that develops over time. (Note: Tolerance to hypoxemia is the reason that “hypoxemic shock” is not a clinical entity.)
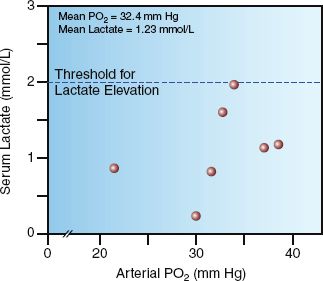
FIGURE 22.1 The relationship between arterial PO2 and blood lactate levels in seven patients with severe hypoxemia (arterial PO2 <40 mm Hg) from acute exacerbation of chronic obstructive lung disease. All lactate levels are within the normal range (≤2 mmol/L), suggesting that severe hypoxemia did not impair tissue oxygenation in these patients. Data from Reference 4.
O2 Therapy and Aerobic Metabolism
Since hypoxemia does not impair aerobic metabolism, then oxygen therapy (which is aimed at relieving hypoxemia) should not be necessary for preserving aerobic metabolism. This is supported by observations like those in Figure 22.2. The graphs in this figure show the effects of breathing 24% and 28% oxygen on the arterial PO2 (PaO2) and systemic oxygen consumption (VO2) in patients with acute exacerbation of chronic ob-structive lung disease. There is a significant increase in the PaO2 with each increment in inhaled O2, but the VO2 remains unchanged. Similar results have been reported in other clinical studies (8,9), and since the VO2 represents the rate of aerobic metabolism, these studies indicate that oxygen therapy does not promote aerobic metabolism.
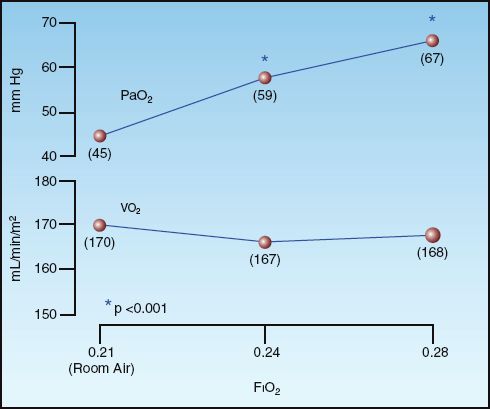
FIGURE 22.2 Acute effects of oxygen therapy on the arterial PO2 (PaO2) and systemic oxygen consumption (VO2) in ICU patients with acute exacerbation of chronic obstructive lung disease. Numbers in parentheses are the mean values for each measurement. FIO2 = fraction of O2 in inhaled gas. Data from Reference 7.
Pure oxygen breathing (a condition known as normobaric hyperoxia) is accompanied by a 10% to 20% decrease in VO2 (10,11), indicating that hyperoxia can inhibit aerobic metabolism(!). This effect has been attributed to oxygen-induced changes in microvascular flow (described next), and it is blocked by the antioxidant N-acetylcysteine (12), indicating that toxic oxygen metabolites are involved.
Oxygen and the Microcirculation
Oxygen therapy produces systemic vasoconstriction (not pulmonary vasoconstriction) that is most pronounced in the small arterioles that control capillary blood flow (12–14). At high concentrations of inhaled O2, the arteriolar constriction can be accompanied by a decrease in functional capillary density (15), and this could result in a decrease in O2 availability in the capillaries. This has been implicated in the reduction of VO2 that occurs during normobaric hyperoxia (described above), and it may also be a mechanism for protecting the tissues from oxygen-induced injury (oxidant injury) during periods of hyperoxia.
Summary and Implications
The information just presented indicates that aerobic metabolism is normally conducted in an oxygen-restricted environment, and this condition persists in the face of severe hypoxemia. Furthermore, attempts to provide more oxygen are met with resistance, as arteriolar vasoconstriction reduces the capillary surface area available for oxygen entry into tissues. Thus, the tissues are normally oxygen-restricted, and it seems they want to remain that way. This is understandable in light of the propensity for oxygen to produce lethal cell damage, as described in the last section of the chapter.
If the scenario just presented is accurate, then oxygen therapy, which does not promote aerobic metabolism, is merely creating an added risk of oxygen-induced (oxidant) damage in the vital organs. The observation that arterial hyperoxia following cardiac arrest is associated with an increased mortality rate (16) is consistent with the notion that oxygen therapy can be dangerous. Considering the prominent role played by oxidant injury in the damaging effects of inflammation (which is a leading cause of morbidity and mortality in the ICU), the current liberal use of oxygen in critically ill patients deserves scrutiny.
OXYGEN DELIVERY SYSTEMS
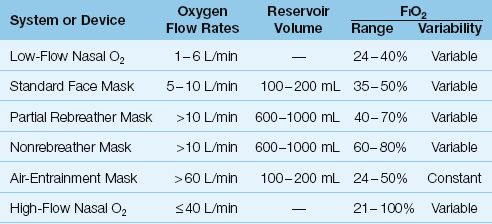
Oxygen delivery systems are classified as low-flow systems, reservoir systems, and high-flow systems (17). Low-flow systems use standard nasal prongs, while reservoir systems use standard face masks and face masks with reservoir bags, and high-flow systems use air-entrainment masks or heated, humidified O2 delivered through nasal prongs. Each system is characterized by the following features: (a) the manner in which the fractional concentration of inspired oxygen (FIO2) is determined, (b) the achievable FIO2 range, (c) the variability of the FIO2, and (d) the type of patient that is most appropriate for the system. The features of different oxygen delivery systems are summarized in Table 22.2
Table 22.2 Oxygen Delivery Systems
Low-Flow Nasal O2
The standard device for low-flow O2 therapy is the nasal cannula or nasal prongs, which deliver oxygen into the nasopharynx at flow rates of 1 to 6 L/min. (The normal inspiratory flow rate during quiet breathing is about 15 L/min (0.25 L/sec), so low-flow nasal O2 represents only a fraction of the inspiratory flow generated by the patient.) A large fraction of the inspired volume is drawn from room air, which means that low-flow nasal O2 does not achieve high concentrations of inhaled oxygen. The FIO2 range during quiet breathing is 24% O2 (at 1 L/min) up to 40% O2 (at 6 L/min), as shown in Table 22.2.
In patients with acute respiratory failure, peak inspiratory flow rates can increase to 30–120 L/min (18). In this situation, low-flow nasal O2 provides an even smaller fraction of the inspiratory flow needs of the patient. For this reason, low-flow nasal O2 is often not adequate for O2 therapy in patients with high ventilatory demands.
Advantages and Disadvantages
The major advantages of nasal prongs are simplicity of use and patient acceptance, including the ability for patients to eat and converse. The major disadvantage is the inability to achieve high concentrations of inhaled O2, particularly in patients with increased ventilatory demands.
Standard Face Masks
Face masks are considered a reservoir system because the mask encloses a volume of 100 to 200 mL. Standard face masks deliver oxygen at flow rates between 5 and 10 L/min; a minimum flow rate of 5 L/min is needed to clear exhaled gas from the mask. Exhalation ports on the side of the face mask also allow room air to be inhaled. This system can achieve a maximum FIO2 of about 60% during quiet breathing.
Advantages and Disadvantages
Standard face masks can provide a slightly higher maximum FIO2 than low-flow nasal prongs, but like nasal prongs, the FIO2 varies with the ventilatory demands of the patient. Face masks are more confining than nasal prongs, and they do not permit oral feeding.
Masks with Reservoir Bags
The addition of a reservoir bag to a standard face mask increases the capacity of the oxygen reservoir by 600 to 1000 mL (depending on the size of the bag). If the reservoir bag is kept inflated, the patient will draw primarily from the gas in the bag. There are two types of reservoir bag devices: partial rebreathers and nonrebreathers.
Partial Rebreather
The device shown in Figure 22.3 is a partial rebreather. This device allows the gas exhaled in the initial phase of expiration to return to the reservoir bag. As exhalation proceeds, the expiratory flow rate declines, and when the expiratory flow rate falls below the oxygen flow rate, exhaled gas can no longer return to the reservoir bag. The initial part of expiration contains gas from the upper airways (anatomic dead space), so the gas that is rebreathed is rich in oxygen and largely devoid of CO2. The patient can inhale room air through the exhalation ports on the mask, but the gas in the reservoir bag is under positive pressure, and inhalation will draw primarily from the gas in the bag. Partial rebreather devices can achieve a maximum FIO2 of about 70%.
Nonrebreather
The device shown in Figure 22.4 is a nonrebreather system. The expiratory ports on the mask are covered with flaps that allow exhaled gas to escape but prevent inhalation of room air gas. There is also a one-way valve between the reservoir bag and the mask that allows inhalation of gas from the bag but prevents exhaled gas from entering the bag (to prevent rebreathing of exhaled gas). Nonrebreather devices can theoretically achieve an FIO2 of 100%, but in reality the maximum FIO2 is closer to 80% (because of leaks around the mask).
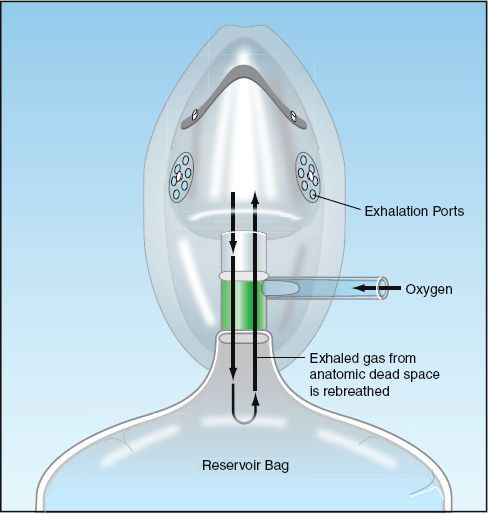
FIGURE 22.3 Partial rebreather system. The initial 100 to 150 mL of exhaled gas (anatomic dead space) is returned to the reservoir bag for rebreathing. Exhaled gas stops entering the reservoir bag when the expiratory flow rate falls below the oxygen flow rate.
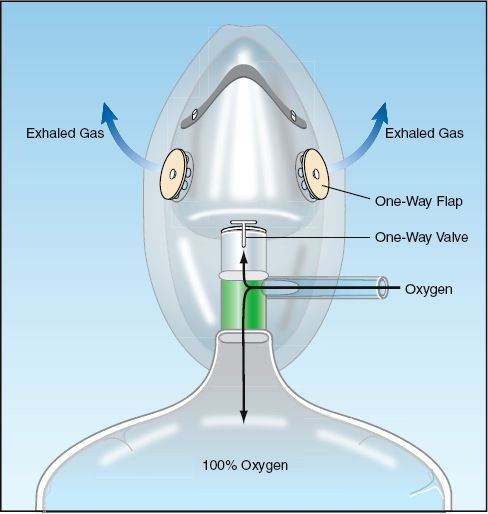
FIGURE 22.4 Nonrebreather system. Flaps on the exhalation ports of the mask prevent inhalation of room air, and a one-way valve between the mask and reservoir bag prevents exhaled gas from entering the bag for rebreathing.
Advantages and Disadvantages
The principal advantage of the reservoir bags is the ability to deliver higher concentrations of inhaled oxygen. The disadvantages are the same as described for face masks. In addition, aerosolized bronchodilator therapy is not possible with reservoir bag devices.
Air Entrainment Device
Air entrainment devices are high-flow systems that deliver a constant FIO2. The operation of an air-entrainment device is shown in Figure 22.5 (19). The end of the oxygen inlet port is narrowed, and this creates a high-velocity stream of gas (analogous to the nozzle on a garden hose). This creates a shearing force known as viscous drag that pulls room air into the device through air-entrainment ports. The greater the flow of O2 into the mask, the greater the volume of air that is entrained, and this keeps the FIO2 constant. The final flow created by the device is in excess of 60 L/min, which exceeds the inspiratory flow rate in most cases of respiratory distress. The FIO2 can be varied by varying the size of the air entrainment port on the device. The FIO2 range of these devices is 24 to 50%.
Note: The mechanism of air entrainment in these devices is known as jet mixing (19). However, air entrainment was originally attributed to the Venturi effect (i.e., where the pressure of a fluid decreases when the fluid flows through a constricted section of a tube), and as a result, masks that used these devices were called Venturi masks or Venti-masks.
Advantages and Disadvantages
The major advantage of air-entrainment devices is the ability to deliver a constant FIO2. This is desirable in patients with chronic CO2 retention, where an inadvertent increase in FIO2 can lead to further increases in arterial PCO2. The major disadvantage of these devices is inability to deliver high concentrations of inhaled O2.
High-Flow Nasal O2
The newest technique of O2 delivery (in adults) is high-flow nasal O2 using heated and humidified gas. Using oxygen that is heated to body temperature and supersaturated with water (to a relative humidity of 99%), O2 flow rates up to 40–60 L/min can be delivered through wide nasal prongs without discomfort and mucosal injury. A commercially available product for high-flow nasal O2 (Vapotherm, Stevensville, MD) allows adjustments for flow rate (1–40 L/min), FIO2 (21–100%), and temperature (usually at 37°C).
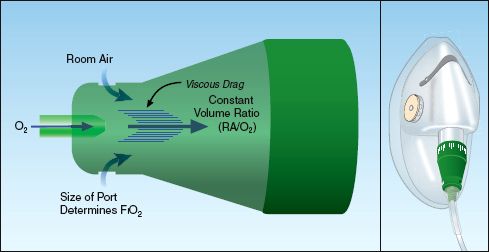
FIGURE 22.5 Function of an air-entrainment device. A narrowing at the oxygen inlet creates a high-velocity stream of gas that creates viscous drag, which pulls in room air (RA). This “jet mixing” keeps the concentration of inhaled oxygen constant, regardless of changes in the flow rate of oxygen. See text for further explanation.
Clinical Experience
The initial experience with high-flow nasal O2 has been very encouraging. Preliminary studies have shown that, in patients who require a high FIO2 using mask devices, switching to humidified, high-flow nasal O2 was associated with a significant improvement in measures of respiratory distress (e.g., reduced respiratory rate, less dyspnea), along with a decrease in the PaO2/FIO2 ratio (indicating improved gas exchange) (20,21). The beneficial effect on gas exchange may be explained by the observation that high-flow nasal O2 creates positive pressure in the nasopharynx (22), which could act like positive end-expiratory pressure (PEEP) to prevent the collapse of alveoli at the end-of expiration (PEEP is described in Chapter 26). The most exciting observation regarding high-flow nasal O2 comes from a study where the improvements resulting from high-flow nasal O2 made it possible to avoid intubation and mechanical ventilation in 75% of the patients studied (21).
Advantages and Disadvantages
The balance sheet for high-flow nasal O2 shows all advantages and no disadvantages (so far). The advantages include improved oxygenation and gas exchange, and a possible role in avoiding intubation and mechanical ventilation in patients with refractory hypoxemia. You will certainly hear more about this promising method of O2 therapy in the near future.
THE TOXIC NATURE OF OXYGEN
Ever wonder why food is stored in vacuum-sealed containers, or why we wrap food in cellophane to keep it fresh? These measures are designed to protect food from exposure to oxygen, which oxidizes and disrupts all organic molecules (including the carbohydrates, proteins, and lipids in food), and is responsible for the decomposition of food. The metabolites of oxygen are even more damaging than the parent molecule, and are capable of inflicting lethal cell injury (23). In fact, contrary to the popular perception that oxygen protects cells from injury in critically ill patients, the accumulating evidence indicates that oxygen (via the production to toxic metabolites) is a source of cell injury in critically ill patients (23–26). The following is a brief description of the toxic nature of oxygen.
Oxygen Metabolism
The metabolism of oxygen takes place at the end of the electron transport chain in mitochondria (within the cytochrome oxidase complex), where the electrons that accumulate as a result of ATP production are cleared by the chemical reduction of oxygen to water. The reaction sequence for this process is shown in Figure 22.6. Oxygen has two unpaired electrons in its outer orbitals with the same directional spin. According to Pauli’s Exclusion Principle (from the Austrian physicist Wolfgang Pauli), no two electrons can occupy the same orbital if they have the same directional spin. This means it is not possible to add an electron pair to oxygen and reduce it to water as a one-step process because one orbital would have two electrons with the same directional spin, which is a quantum impossibility. Because of this spin restriction, oxygen is metabolized in a series of single-electron reduction reactions, and this produces some highly reactive intermediates.
The intermediates in oxygen metabolism include the superoxide radical, hydrogen peroxide, and the hydroxyl radical. The behavior of these metabolites is summarized in the following statements (23):
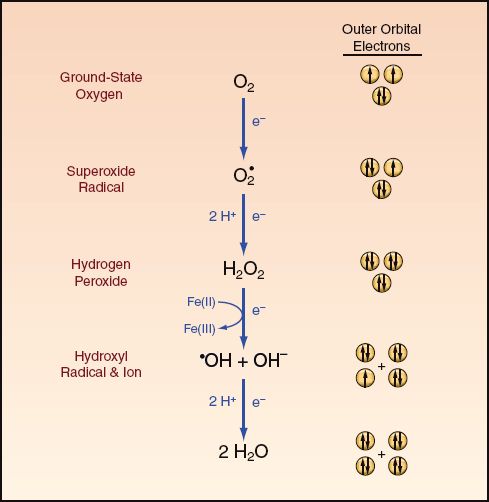
FIGURE 22.6 The metabolism of molecular oxygen to water involves a series of four single-electron reduction reactions. Orbital diagrams on the right side of the figure show the directional spin of electrons in the outer orbitals of each reactant. Free radical metabolites are indicated by a superscripted dot. See text for explanation.
1. All the intermediates in O2 metabolism are oxidizing agents or oxidants that are capable of disrupting and damaging vital cell components such as membrane lipids, cytoplasmic proteins, and nuclear DNA.
2. The superoxide and hydroxyl radicals are free radicals (i.e., they have an unpaired electron in their outer orbitals), and free radicals tend to be highly reactive. (Oxygen is also a free radical, but it is only sluggishly reactive because of the spin restriction just described.)
3. The hydroxyl radical is the most reactive molecule known in biochemistry, and enters into a reaction within three molecular diameters of its point of origin (23)! It is the most destructive O2 metabolite, and is a major source of oxygen-induced cell injury.
4. Free iron in its reduced form (Fe++) catalyses the formation of the hydroxyl radical, and thus free iron can act as a pro-oxidant (see later).
5. Hydrogen peroxide is not a free radical, and is the least reactive of the O2 metabolites. This low reactivity allows hydrogen peroxide to move freely throughout the body, creating the potential for widespread oxidant injury.
Normally, at least 95% of oxygen is completely reduced to water, and only 3–5% of O2 metabolism generates the damaging O2 metabolites (27). Depletion of selected antioxidants like glutathione will change this proportion (see later).
Neutrophil Activation
Oxygen metabolites play a major role in the inflammatory response, as described in Chapter 14 (see pages 263–266). The activation of neutrophils involves a marked (up to 50-fold) increase in cellular O2 consumption (26). This is known as the respiratory burst, and its purpose is not to generate high-energy ATP, but to produce toxic O2 metabolites, which are stored in cytoplasmic granules. Oxygen metabolism in neutrophils also generates hypochlorite (see Figure 14.1 on page 265), which is highly microbicidal (and is the active ingredient in household bleach).
When neutrophils arrive at the sight of infection, they degranulate and release the stored O2 metabolites to damage and destroy the invading microbes. Unfortunately, the O2 metabolites can also damage the tissues of the host if adequate antioxidant protection is not available (25,26).
Chain Reactions
The damage produced by O2 metabolites is magnified because of the tendency for free radicals to create chain reactions (28). When a free radical reacts with a non-radical, the non-radical loses an electron and is transformed into a free radical, which can then remove an electron from another nonradical to produce another free radical, and so on. This creates a self-sustaining reaction that continues after the inciting event is eliminated. A fire is a familiar example of a chain reaction involving free radicals. Chain reactions would also explain the progression of inflammatory multiorgan failure in severe sepsis and septic shock after the infection has been eradicated.
Antioxidant Protection
Oxidative (oxygen-related) injury is kept in check by a vast array of endogenous antioxidants (i.e., atoms or molecules that prevent or block the actions of oxidizing agents.) The following is a brief description of the major antioxidants and their possible role in critical illness.
Superoxide Dismutase
Superoxide dismutase (SOD in Figure 22.7) is an enzyme that facilitates the conversion of superoxide radicals to hydrogen peroxide. Although SOD is considered essential for aerobic life (27), its role in critical illness is unclear, and it can act as a pro-oxidant as well as an antioxidant (27,29). The pro-oxidant effect may be the result of hydrogen peroxide production.
Glutathione
Glutathione is a sulfur-containing tripeptide that is considered the major intracellular antioxidant in the human body (29,30). In its reduced form, glutathione (GSH in Figure 22.7) donates electrons to reduce hydrogen peroxide to water in a reaction that is catalyzed by a selenium-dependent enzyme, glutathione peroxidase (Se-GPx in Figure 22.7):
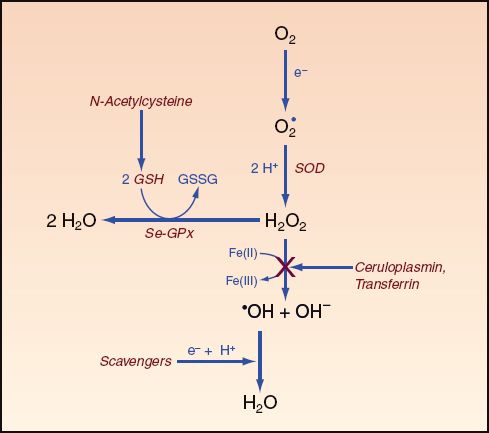
FIGURE 22.7 Actions of endogenous and exogenous antioxidants (highlighted in red). SOD = superoxide dismutase, Se-GPx = selenium-glutathione peroxidase complex, GSH = reduced glutathione, GSSG = oxidized glutathione (a dipeptide connected by a disulfide bridge). See text for explanation.
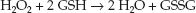 (22.2)
(22.2)
Glutathione is present in high concentrations is most mammalian cells (0.5–10 mM/L), and is synthesized de novo within cells. It can be ex-ported extracellularly, but plasma levels are three orders of magnitude lower than intracellular levels (31). However, glutathione levels in lung lavage fluid are 140-fold higher than plasma (32), suggesting that glutathione plays an important role in protecting the lung from oxidant injury. There is evidence that intracellular glutathione is depleted in critically ill patients (33).
N-ACETYLCYSTEINE: Glutathione does not move readily into cells, and exogenous glutathione administration has little effect on intracellular levels (34). However, the popular mucolytic agent N-acetylcysteine is a glutathione analogue that can cross cell membranes and serve as a glutathione surrogate (which is the mechanism for the beneficial effects of N-acetylcysteine in acetaminophen toxicity, as described in Chapter 54). N-acetylcysteine has been used sparingly as an antioxidant, but there have been some favorable results (35).
SELENIUM: Selenium is an essential trace element that serves as a cofactor for the glutathione peroxidase enzyme in humans. The recommended dietary allowance (RDA) for selenium is 55 micrograms daily in adult men and women (36). The absence of dietary selenium produces measurable decreases in glutathione peroxidase activity after just one week (37). Selenium levels in blood are typically low in critically ill patients (38), and high-dose selenium replacement (1000 ∝g IV daily) has been associated with improved survival in patients with severe sepsis and septic shock (38).
Selenium status can be monitored using whole blood selenium (normal range = 0.96–1.78 ∝mol/L) or serum selenium (normal range=0.72–1.33 ∝mol/L) (33). If needed, selenium can be given intravenously as sodium selenite. The highest daily dose that is considered safe is 200 micrograms.
Vitamin E
Vitamin E (alpha-tocopherol) is a lipid-soluble vitamin that is found in the interior of most cell membranes, where it serves as a “chain breaking” antioxidant to halt the progression of lipid peroxidation, which proceeds as a chain reaction. To accomplish this, vitamin E donates an electron to a free radical intermediate in lipid peroxidation, and in so doing, vitamin E becomes a free radical, but an innocuous one. (Note: Lipid peroxidation is the oxidation of polyunsaturated fatty acids and is known as rancidity when it occurs in food products.)
Vitamin E depletion has been reported in patients with acute respiratory distress syndrome (39), and there is evidence that high-dose vitamin E has beneficial effects in trauma victims (see later). The normal concentration of vitamin E in plasma is 1 mg/dL, and a level below 0.5 mg/dL is evidence of deficiency (40).
Vitamin C
Vitamin C (ascorbic acid) is a water-soluble antioxidant that operates primarily in the extracellular space. The importance of vitamin C as an antioxidant is unclear, but it is a powerful reducing agent that can serve as a scavenger (i.e., donate electrons) for the superoxide and hydroxyl radicals (30). High-dose vitamin C has shown some promise in reducing pulmonary complications in trauma patients (see later).
Caeruloplasmin and Transferrin
Ceruloplasmin and transferrin account for most of the antioxidant activity in plasma (41). The antioxidant activity of both proteins is related to their actions in limiting free iron in the reduced form, Fe (II), which will limit the production of the hydroxyl radical. Caeruloplasmin oxidizes iron from the Fe (II) to the Fe (III) state, and transferrin binds iron in the oxidized or Fe (III) state. The role of free iron as a pro-oxidant (42) may explain why most of the iron in the body is bound to proteins or sequestered (e.g., in the bone marrow).
Oxidant Stress
The risk of oxidant injury is determined by the balance between oxidant and antioxidant activities. When oxidant activity exceeds the neutralizing capacity of the antioxidants, the excess or unopposed oxidation can promote tissue injury. This condition of unopposed biological oxidation is known as oxidant stress. Unfortunately, there is no clinical measure of oxidant stress, so the presence of this condition must be inferred.
Pulmonary Oxygen Toxicity
Pulmonary oxygen toxicity is described as an inflammatory lung injury (similar to the acute respiratory distress syndrome described in the next chapter) that occurs in patients who have inhaled gas with an FIO2 >60% for longer than 48 hours. This description, however, does not take into account the information that follows.
Species Differences
The tendency to develop pulmonary oxygen toxicity varies in different species. For example, laboratory rats will die of respiratory failure after 5 to 7 days of breathing 100% O2, while sea turtles can breathe pure O2 indefinitely without harm (43). This species-specific effect is important because experimental studies of pulmonary oxygen toxicity have been conducted almost solely in laboratory rats. There is limited information about the tendency of humans to develop pulmonary oxygen toxicity.
HUMAN STUDIES: In healthy volunteers, inhalation of 100% O2 for 6 to 12 hours results in a tracheobronchitis and a decrease in vital capacity attributed to absorption atelectasis (44). Prolonged exposure to 100% O2 has been reported in only 6 humans: 5 with irreversible coma who received 100% O2 for 3 to 4 days (45), and one healthy volunteer who inhaled pure O2 for 4.5 days (46). In all these cases, the subjects developed a pulmonary condition that was consistent with inflammatory lung injury.
What FIO2 Is Toxic?
Based on the observation of a decreased vital capacity when the FIO2 exceeds 60% (44), the threshold FIO2 for pulmonary O2 toxicity was set at 60%. However, adopting a single threshold FIO2 for all patients neglects the contribution of endogenous antioxidants to the risk of oxygen toxicity. If the antioxidant stores in the lungs are depleted, oxygen toxicity can be expected at FIO2 levels below 60%. Since antioxidant depletion seems to be common in ICU patients (33,38,39), it is reasonable to assume that any FIO2 above 21% (room air) can represent a toxic exposure to oxygen in critically ill patients. Therefore, the best practice is to reduce the FIO2 to the lowest tolerable level; e.g., the lowest FIO2 needed to maintain an arterial O2 saturation ≥90%.
Promoting Antioxidant Protection
There is no clinical measure of pulmonary O2 toxicity. However, considering the evidence of antioxidant depletion in critically ill patients (33,38,39), antioxidant supplementation is a reasonable consideration for reducing the risk of pulmonary oxygen toxicity. Evidence of benefit from such an approach is provided by a study in trauma patients (47), which showed that a high-dose antioxidant cocktail of vitamin C (1000 mg every 8 hrs), vitamin E (1000 units every 8 hrs), and selenium (200 ∝g daily) for 7 days was associated with a significant decline in cases of respiratory failure and ventilator dependence (47).
Antioxidant supplementation could be guided by available measures of antioxidant protection (e.g., serum levels of selenium), or it could be used routinely in patients considered at risk for pulmonary O2 toxicity.
A FINAL WORD
Why Is Oxygen a Vasoconstrictor?
For those who like teleological explanations for biological design, the observation that oxygen is an arteriolar vasoconstrictor merits some attention. As shown in the first section of the chapter, aerobic metabolism is normally conducted in an oxygen—restricted environment; i.e., most of the O2 in the body is attached to hemoglobin, while very little is present in tissues because O2 does not readily dissolve in water. Further-more, attempts to enhance tissue oxygenation with O2 therapy are thwarted because of oxygen’s actions as an arteriolar vasoconstrictor, which reduces functional capillary flow. Thus, the body resists attempts to increase tissue oxygenation, and the likely purpose of this design is to limit the risk of oxygen-induced tissue injury. In summary, it seems the human body is designed to maintain an oxygen-restricted environment in the tissues to limit the risk of oxygen-induced cell injury. If this is the case, then the liberal use of oxygen therapy in critically ill patients deserves re-evaluation.
REFERENCES
Books
Lane N. Oxygen: The Molecule that Made the World. New York: Oxford University Press, 2002.
Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. 4th ed, New York: Oxford University Press, 2007.
Banerjee R (ed). Redox Biochemistry. Hoboken, NJ: Wiley & Sons, 2008.
Insights
1. Whalen WJ, Riley J. A microelectrode for measurement of intracellular PO2. J Appl Physiol 1967; 23:798–801.
2. Sair M, Etherington PJ, Winlove CP, Evans TW. Tissue oxygenation and perfusion in patients with systemic sepsis. Crit Care Med 2001; 29:1343–1349.
3. American Association for Respiratory Care. Clinical practice guideline: oxygen therapy for adults in the acute care facility. Respir Care 2002; 47:717–720.
4. Eldridge FE. Blood lactate and pyruvate in pulmonary insufficiency. N Engl J Med 1966; 274:878–883.
5. Lundt T, Koller M, Kofstad J. Severe hypoxemia without evidence of tissue hypoxia in the adult respiratory distress syndrome. Crit Care Med 1984; 12:75–76.
6. Abdelsalam M. Permissive hypoxemia. Is it time to change our approach? Chest 2006; 129:210–211.
7. Lejeune P, Mols P, Naeije R, et al. Acute hemodynamic effects of controlled oxygen therapy in decompensated chronic obstructive pulmonary disease. Crit Care Med 1984; 12:1032–1035.
8. Corriveau ML, Rosen BJ, Dolan GF. Oxygen transport and oxygen consumption during supplemental oxygen administration in patients with chronic obstructive pulmonary disease. Am J Med 1989; 87:633–636.
9. Esteban A, Cerde E, De La Cal MA, Lorente JA. Hemodynamic effects of oxygen therapy in patients with acute exacerbations of chronic obstructive pulmonary disease. Chest 1993; 104:471–475.
10. Reinhart K, Bloos F, Konig F, et al. Reversible decrease of oxygen consumption by hyperoxia. Chest 1991; 99:690-694.
11. Lauscher P, Lauscher S, Kertscho H, et al. Hyperoxia reversibly alters oxygen consumption and metabolism. Scientific World Journal, volume 2012, article ID 410321, 2012. (An open access article, accessed at www.PubMed.com on 12/24/2012.)
12. Packer M, Lee WH, Medina N, Yushak M. Systemic vasoconstrictor effects of oxygen administration in obliterative pulmonary vascular disorders. Am J Cardiol 1986; 57:853–858.
13. Bongard O, Bounameaux H, Fagrell B. Effects of oxygen on skin microcirculation in patients with peripheral arterial occlusive disease. Circulation 1992; 86:878–886.
14. Duling BR. Microvascular responses to alterations in oxygen tension. Circ Res 1972; 31:481-489.
15. Tsai AG, Cabrales P, Winslow RM, Intaglietta M. Microvascular oxygen distribution in awake hamster window chamber model during hyperoxia. Am J Physiol 2003; 285:H1537–H1545.
16. Kilgannon JH, Jones AE, Shapiro NI, et al. Association between arterial hyperoxia following resuscitation from cardiac arrest and in-hospital mortality. JAMA 2010; 303:2165–2171.
Oxygen Delivery Systems
17. Heuer AJ, Scanlan CL. Medical gas therapy. In Wilkins RL, Stoller JK, Kacmarek RM, eds. Egan’s Fundamentals of Respiratory Care, 9th ed, St. Louis: Mosby Elsevier, 2009.
18. L’Her E, Deye N, Lellouche F, et al. Physiologic effects of noninvasive ventilation during acute lung injury. Am J Respir Crit Care Med 2005; 172:1112-1118.
19. Scacci R. Air entrainment masks: jet mixing is how they work. The Bernoulli and Venturi principles is how they don’t. Respir Care 1979; 24:928–931.
20. Roca O, Riera J, Torres F, Masclans JR. High-flow oxygen therapy in acute respiratory failure. Respir Care 2010; 55:408–413.
21. Sztrymf B, Messika J, Bertrand F, et al. Beneficial effects of humidified high flow nasal oxygen in critical care patients: a prospective pilot study. Inten-sive Care Med 2011; 37:1780–1786.
22. Parke R, MacGuinness S, Eccleston M. Nasal high-flow therapy delivers low-level positive airway pressure. Br J Anesth 2009; 103:886–890.
The Toxic Nature of Oxygen
23. Halliwell B, Gutteridge JMC. Oxygen is a toxic gas – an introduction to oxygen toxicity and reactive oxygen species. In Free Radicals in Biology and Medicine. 4th ed. New York: Oxford University Press, 2007:1–28.
24. Alonso de Vega JM, Diaz J, Serrano E, Carbonell LF. Oxidative stress in critically ill patients with systemic inflammatory response syndrome. Crit Care Med 2002; 1782–1788.
25. Fink M. Role of reactive oxygen and nitrogen species in acute respiratory distress syndrome. Curr Opin Crit Care 2002; 8:6–11.
26. Anderson BO, Brown JM, Harken A. Mechanisms of neutrophil-mediated tissue injury. J Surg Res 1991; 51:170–179.
27. Michiels C, Raes M, Toussant O, Remacle J. Importance of Se-Glutathione, peroxidase, catalase, and CU/ZN-SOD for cell survival against oxidative stress Free Rad Biol Med 1994; 17:235–248.
28. Halliwell B, Gutteridge JMC. The chemistry of free radicals and related ‘reactive species’. In: Free Radicals in Biology and Medicine. 4th ed. New York: Oxford University Press, 2007:30–79.
29. Halliwell B, Gutteridge JMC. Antioxidant defenses: endogenous and diet derived. In: Free Radicals in Biology and Medicine, 4th ed. New York: Oxford University Press, 2007:79–185.
30. Suttorp N, Toepfer W, Roka L. Antioxidant defense mechanisms of endothelial cells:glutathione redox cycle versus catalase. Am J Physiol 1986; 251:C671–C680.
31. Cantin AM, Begin R. Glutathione and inflammatory disorders of the lung. Lung 1991; 169:123–138.
32. Cantin AM, North SI, Hubbard RC, Crystal RG. Normal alveolar epithelial lining fluid contains high levels of glutathione. J Appl Physiol 1987; 63:152–157.
33. Hammarqvist F, Luo JL, Cotgreave IA, et al. Skeletal muscle glutathione is depleted in critically ill patients. Crit Care Med 1997; 25:78–84.
34. Robinson M, Ahn MS, Rounds JD, et al. Parenteral glutathione monoester enhances tissue antioxidant stores. J Parent Ent Nutrit 1992; 16:413–418.
35. Suter PM, Domenighetti G, Schaller MD, et al. N-acetylcysteine enhances recovery from acute lung injury in man: a randomized, double-blind, placebo-controlled clinical study. Chest 1994; 105:190–194.
36. Institute of Medicine, Food and Nutrition Board. Dietary reference intakes: vitamin C, vitamin E, selenium, and carotenoids. Washington, DC: National Academy Press, 2000.
37. Sando K, Hoki M, Nezu R, et al. Platelet glutathione peroxidase activity in long-term total parenteral nutrition with and without selenium supplementation. J Parent Ent Nutrit 1992; 16:54–58.
38. Angstwurm MWA, Engelmann L, Zimmerman T, et al. Selenium in Intensive Care (SIC): results of a prospective randomized, placebo-controlled, multiple-center study in patients with severe systemic inflammatory response syndrome, sepsis, and septic shock. Crit Care Med 2007; 35:118–126.
39. Pincemail J, Bertrand Y, Hanique G, et al. Evaluation of vitamin E deficiency in patients with adult respiratory distress syndrome. Ann NY Acad Sci 1989; 570:498–500.
40. Meydani M. Vitamin E. Lancet 1995; 345:170–176.
41. Halliwell B, Gutteridge JMC. Role of free radicals and catalytic metal ions in human disease. Methods Enzymol 1990; 186:1–85.
42. Herbert V, Shaw S, Jayatilleke E, Stopler-Kasdan T. Most free-radical injury is iron-related: it is promoted by iron, hemin, haloferritin and vitamin C, and inhibited by desferrioxamine and apoferritin. Stem Cells 1994; 12:289–303.
Pulmonary Oxygen Toxicity
43. Fanburg BL. Oxygen toxicity: why can’t a human be more like a turtle? Intensive Care Med 1988; 3:134–136.
44. Lodato RF. Oxygen toxicity. Crit Care Clin 1990; 6:749–765.
45. Barber RE, Hamilton WK. Oxygen toxicity in man. N Engl J Med 1970; 283:1478–1483.
46. Winter PM, Smith G. The toxicity of oxygen. Anesthesiology 1972; 37:210–212.
47. Dossett GAM, Fleming SB, Abumrad NN, Cotton BA. High-dose antioxidant administration is associated with a reduction in post-injury complications in critically ill trauma patients. Injury 2011; 42:78–82.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


