Opioids produce analgesia as well as serious side effects. All physicians who prescribe opioids for relief of acute or chronic pain need to know how to use these drugs safely. This requires an in-depth understanding of the pharmacokinetics and pharmacodynamics of opioids as well as the acquisition of sufficient clinical experience in their use.
 Opioids act through specific opioid receptors on neuronal tissues such as peripheral nerves and neurons in the spinal cord and brain. The most important receptors include the μ-opioid receptor (MOR), δ-opioid receptor (DOR), and κ-opioid receptor (KOR). For anesthesia and pain relief, the MOR is most important. Opioids also act through non-neuronal pathways, such as those affecting the immune system, which may be of relevance in the treatment of inflammatory pain.
Opioids act through specific opioid receptors on neuronal tissues such as peripheral nerves and neurons in the spinal cord and brain. The most important receptors include the μ-opioid receptor (MOR), δ-opioid receptor (DOR), and κ-opioid receptor (KOR). For anesthesia and pain relief, the MOR is most important. Opioids also act through non-neuronal pathways, such as those affecting the immune system, which may be of relevance in the treatment of inflammatory pain.
 Opioids can be classified according to the strength or potency based on the plasma concentrations at which they exert their effects (C50 or the plasma concentration causing a 50% effect). Strong opioids include fentanyl, sufentanil, and remifentanil. Weak opioids include codeine and tramadol. An intermediate group includes morphine, methadone, oxycodone, and buprenorphine.
Opioids can be classified according to the strength or potency based on the plasma concentrations at which they exert their effects (C50 or the plasma concentration causing a 50% effect). Strong opioids include fentanyl, sufentanil, and remifentanil. Weak opioids include codeine and tramadol. An intermediate group includes morphine, methadone, oxycodone, and buprenorphine.
 Endogenous opioid pathways are activated in cases of stress-induced analgesia, placebo-induced analgesia, and conditioning pain modulation (CPM). CPM occurs when pain arising from one focus is decreased by application of a second painful stimulus.
Endogenous opioid pathways are activated in cases of stress-induced analgesia, placebo-induced analgesia, and conditioning pain modulation (CPM). CPM occurs when pain arising from one focus is decreased by application of a second painful stimulus.
 Opioid-induced hyperalgesia (OIH) is a paradoxical opioid effect whereby pain sensitivity increases during or following escalating opioid treatment. It is also observed postoperatively following the use of a remifentanil infusion during anesthesia. Greater and more frequent doses of morphine are required to treat postoperative pain. OIH may be treated and prevented by administration of a low dose of ketamine, an N-methyl-D-aspartate (NMDA) receptor antagonist.
Opioid-induced hyperalgesia (OIH) is a paradoxical opioid effect whereby pain sensitivity increases during or following escalating opioid treatment. It is also observed postoperatively following the use of a remifentanil infusion during anesthesia. Greater and more frequent doses of morphine are required to treat postoperative pain. OIH may be treated and prevented by administration of a low dose of ketamine, an N-methyl-D-aspartate (NMDA) receptor antagonist.
 The pharmacokinetic characteristics of a drug determine its behavior in the body of a patient. One important pharmacokinetic concept is that of context-sensitive half-time (CSt½). This is the time needed for the drug’s plasma concentration to decrease by 50% from a steady-state concentration. For most opioids, this value is dependent on the duration of drug infusion. For example, for fentanyl the CSt½ increases rapidly with the infusion duration. In contrast, the CSt½ of remifentanil is independent of the infusion duration due to its rapid elimination from plasma. Thus it remains stable at about 2 minutes.
The pharmacokinetic characteristics of a drug determine its behavior in the body of a patient. One important pharmacokinetic concept is that of context-sensitive half-time (CSt½). This is the time needed for the drug’s plasma concentration to decrease by 50% from a steady-state concentration. For most opioids, this value is dependent on the duration of drug infusion. For example, for fentanyl the CSt½ increases rapidly with the infusion duration. In contrast, the CSt½ of remifentanil is independent of the infusion duration due to its rapid elimination from plasma. Thus it remains stable at about 2 minutes.
 Opioid metabolism is affected by drugs that interfere with metabolizing cytochrome P450 enzymes, most importantly CYP3A4. Opioid metabolites can be active or inactive. Active metabolites need to be considered when treating patients. For example, the active metabolite of morphine, morphine-6-glucuronide, can accumulate in patients with renal impairment. Genetic variability in the CYP system can have important clinical implications, especially in case of variations in the copies of a gene coding for the metabolizing enzyme. An example is the enzyme CYP2D6 which catalyzes the conversion of codeine into morphine. Patients with multiple copies of the CYP2D6 gene and who receive codeine will have large plasma morphine concentrations with all related effects and side effects.
Opioid metabolism is affected by drugs that interfere with metabolizing cytochrome P450 enzymes, most importantly CYP3A4. Opioid metabolites can be active or inactive. Active metabolites need to be considered when treating patients. For example, the active metabolite of morphine, morphine-6-glucuronide, can accumulate in patients with renal impairment. Genetic variability in the CYP system can have important clinical implications, especially in case of variations in the copies of a gene coding for the metabolizing enzyme. An example is the enzyme CYP2D6 which catalyzes the conversion of codeine into morphine. Patients with multiple copies of the CYP2D6 gene and who receive codeine will have large plasma morphine concentrations with all related effects and side effects.
 Opioid effects are variable among patients. Dosing is optimum when opioids are titrated to the effect. It is also important to take into account the delay between the administration of an opioid and its effect, which is defined as the blood–effect-site equilibration half-life or t½ke0. This will allow proper and timely dosing particularly when anticipating a stressful event (laryngoscopy, intubation, skin incision, etc.) and administering opioids to prevent the occurrence of a large hemodynamic response to these stimuli. The t½ke0 of morphine is 1 to 2 hours! Thus it is important to give an initial bolus dose or morphine 30 to 45 minutes before the end of surgery when using the drug for postoperative pain relief.
Opioid effects are variable among patients. Dosing is optimum when opioids are titrated to the effect. It is also important to take into account the delay between the administration of an opioid and its effect, which is defined as the blood–effect-site equilibration half-life or t½ke0. This will allow proper and timely dosing particularly when anticipating a stressful event (laryngoscopy, intubation, skin incision, etc.) and administering opioids to prevent the occurrence of a large hemodynamic response to these stimuli. The t½ke0 of morphine is 1 to 2 hours! Thus it is important to give an initial bolus dose or morphine 30 to 45 minutes before the end of surgery when using the drug for postoperative pain relief.
 Opioids reduce the requirement of inhalational anesthetics and propofol during anesthesia, which makes rapid awakening from anesthesia possible. Using known pharmacokinetic and pharmacodynamic data it can be determined what doses and plasma concentrations will permit the shortest time to awakening. For example, termination of drug infusions at plasma concentrations of propofol 1.5 μg/mL and remifentanil 9.0 ng/mL will lead to patient awakening within 6.5 minutes.
Opioids reduce the requirement of inhalational anesthetics and propofol during anesthesia, which makes rapid awakening from anesthesia possible. Using known pharmacokinetic and pharmacodynamic data it can be determined what doses and plasma concentrations will permit the shortest time to awakening. For example, termination of drug infusions at plasma concentrations of propofol 1.5 μg/mL and remifentanil 9.0 ng/mL will lead to patient awakening within 6.5 minutes.
 Administration of opioids can potentially lead to life-threatening respiratory depression. The incidence of serious perioperative respiratory events is approximately 0.5% (1 in every 200 patients). Respiratory problems from opioids are more common in special patient populations including patients with obstructive sleep apnea (OSA), premature neonates, and very old or very ill patients. Careful, slow, infusion of opioids allows the gradual accumulation of arterial CO2 which serves as a respiratory stimulant at the chemoreceptors and lowers the probability of apnea.
Administration of opioids can potentially lead to life-threatening respiratory depression. The incidence of serious perioperative respiratory events is approximately 0.5% (1 in every 200 patients). Respiratory problems from opioids are more common in special patient populations including patients with obstructive sleep apnea (OSA), premature neonates, and very old or very ill patients. Careful, slow, infusion of opioids allows the gradual accumulation of arterial CO2 which serves as a respiratory stimulant at the chemoreceptors and lowers the probability of apnea.
 The nonspecific opioid receptor antagonist, naloxone, is the drug of choice to reverse opioid-induced respiratory depression. The required dose of naloxone depends on the pharmacokinetic and pharmacodynamic properties and the dose of the opioid that needs reversal. Postoperatively, when there is persistent apnea, opioid concentrations are often just above the threshold for respiratory depression. Intravenous administration of naloxone using incremental doses of 40 to 80 μg, to a cumulative dose of less than 400 μg, may be sufficient for breathing to resume.
The nonspecific opioid receptor antagonist, naloxone, is the drug of choice to reverse opioid-induced respiratory depression. The required dose of naloxone depends on the pharmacokinetic and pharmacodynamic properties and the dose of the opioid that needs reversal. Postoperatively, when there is persistent apnea, opioid concentrations are often just above the threshold for respiratory depression. Intravenous administration of naloxone using incremental doses of 40 to 80 μg, to a cumulative dose of less than 400 μg, may be sufficient for breathing to resume.
 Apart from respiratory depression, opioids cause a large number of side effects that require attention: Nausea with or without vomiting, smooth muscle spasms, skeletal muscle rigidity (fentanyl and congeners), histamine release (morphine, codeine, meperidine), itching (especially after spinal administration), miosis, sedation, and dizziness. The cardiovascular side effects of opioids include bradycardia and hypotension but are generally mild at usual clinical doses. However, when combined with anesthetics, even at usual clinical doses, or in severely ill patients, opioids may produce hemodynamic instability which requires treatment.
Apart from respiratory depression, opioids cause a large number of side effects that require attention: Nausea with or without vomiting, smooth muscle spasms, skeletal muscle rigidity (fentanyl and congeners), histamine release (morphine, codeine, meperidine), itching (especially after spinal administration), miosis, sedation, and dizziness. The cardiovascular side effects of opioids include bradycardia and hypotension but are generally mild at usual clinical doses. However, when combined with anesthetics, even at usual clinical doses, or in severely ill patients, opioids may produce hemodynamic instability which requires treatment.
Multimedia
 Dose-Effect Curves
Dose-Effect Curves
 Drug Concentration and Effect
Drug Concentration and Effect
 Vomiting Pharmacology
Vomiting Pharmacology
INTRODUCTION
Administration of large doses of opioids has traditionally been restricted to anesthesiologists and other anesthesia providers because of their extensive experience and expertise in the use of these potent analgesics during anesthesia. Recently, however, there has been an exponential increase in the prescription of high-dose opioids by other healthcare providers for treatment of patients with chronic non-cancer pain (3% of adult US population).1 This “epidemic” of opioid use by nonsurgical patients, coupled with an emphasis on aggressive and effective postoperative pain management for patients undergoing surgery, has resulted in increasingly complex postoperative pain management problems for surgical patients and  an increase in opioid-related complications for patients with pain in general.2,3 Consequently, expertise in the use of opioids is not only required in the operating room and postoperatively, but also when caring for patients with chronic pain in a nonsurgical setting. Certainly, knowledge on the safe use of opioids is a conditio sine qua non for all anesthesiologists and other anesthesia providers. An in-depth knowledge of the pharmacokinetics (PK), the pharmacodynamics (PD), and the side effect profile associated with different opioids, dosing regimens, and routes of administration is essential for their safe intra- and postoperative use. This chapter will review the endogenous opioid system, discuss the PK and PD properties of opioids relevant to perioperative dosing strategies, and delineate the associated side effect profiles of exogenous opioid analgesics.
an increase in opioid-related complications for patients with pain in general.2,3 Consequently, expertise in the use of opioids is not only required in the operating room and postoperatively, but also when caring for patients with chronic pain in a nonsurgical setting. Certainly, knowledge on the safe use of opioids is a conditio sine qua non for all anesthesiologists and other anesthesia providers. An in-depth knowledge of the pharmacokinetics (PK), the pharmacodynamics (PD), and the side effect profile associated with different opioids, dosing regimens, and routes of administration is essential for their safe intra- and postoperative use. This chapter will review the endogenous opioid system, discuss the PK and PD properties of opioids relevant to perioperative dosing strategies, and delineate the associated side effect profiles of exogenous opioid analgesics.
Short History
Opium is among the oldest drugs in the world. Fossilized opium poppies have been found in Neanderthal excavation sites dating back to 30,000 BC. Many old civilizations, including the Sumerians, Egyptians, Greeks, Romans, and Chinese, used opium for nutritional, medicinal, euphoric, spiritual, and religious purposes. The first written reference to the medicinal use of the opium poppy is described in a Sumerian text dated near 4,000 BCE. Just over 200 years ago, the German pharmacist and chemist Friedrich Sertürner isolated a stable alkaloid crystal from the opium sap and named it morphine after the Greek god of dreams, Morpheus.4,5 Morphine was 10-fold more potent than opium and soon replaced it not only for the treatment of severe pain, but also for a myriad of other purposes such as cough and diarrhea. After the invention of the hypodermic syringe in the 1850s, the Englishman Alexander Wood was the first to inject morphine in a controlled fashion into a patient producing more than a day’s sleep (1853).6 The first reported casualty from morphine occurred shortly thereafter when Wood injected his wife with morphine resulting in a fatal overdose from respiratory depression.6 Morphine revolutionized the treatment of the wounded in battlefield medicine, but euphoric and addictive properties led to the addiction of thousands of soldiers to morphine during the American Civil War.
The synthesis of heroin in 1874 was based on the empirical finding that boiling morphine with specific acids caused the replacement of the two morphine –OH groups by –OCOH3 producing diamorphine or heroin (Fig. 19-1).
FIGURE 19-1. Chemical structures of common opioids.
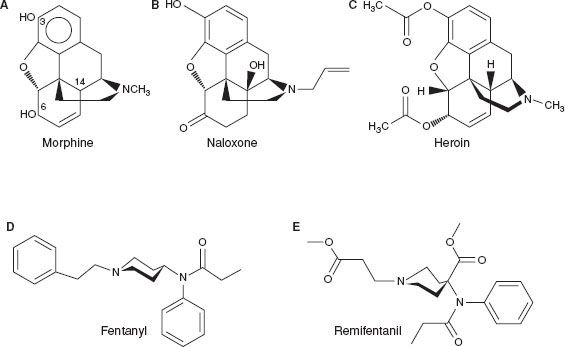
After the structure of morphine was determined in the 1920s, the synthesis of new morphine-like opioid compounds was based on chemical principles rather than empirical discoveries. In 1937, meperidine (or pethidine) became the first synthetic opioid synthesized on the basis of the central structure of morphine. Since then many synthetic and semisynthetic opioids have been produced, including the clinically important opioid antagonists naloxone and naltrexone, by replacing the N-methyl substituent in morphine with allyl and cyclopropylmethyl groups, respectively (Fig. 19-1).7
For clinical use during anesthesia the most important opioids are the piperidines fentanyl, sufentanil, alfentanil, and remifentanil. These opioids produce potent analgesia and suppression of cardiovascular responses to noxious stimulation from surgery with predictable PK and PD. The latest potent opioid to be registered is tapentadol.8,9 This molecule acts via activation of the opioid receptors at spinal and supraspinal sites and by inhibition of the reuptake of norepinephrine, activating α2-adrenergic receptors in the spinal cord dorsal horn. This produces a synergistic analgesic effect with a more desirable gastrointestinal (GI) side effect profile. The continued development of opioids with complex simultaneous actions at opioid and non-opioid target sites is facilitated by new information gained about mechanisms involved in endogenous pain and analgesia as well as advances in pain-related pharmacology. Development of new opioids is also driven by concerns that the side effect profile of potent opioids, which presents a serious risk to patients, needs to be minimized.
The Endogenous Opioid System
A major breakthrough in the understanding of opioid pharmacology came from a series of discoveries of opioid receptors, endogenous opioid peptides, their encoding genes, and endogenous opioid alkaloids. The endogenous opioid system is composed of a family of structurally related endogenous peptides that act at a four-member opioid receptor family consisting of the μ-opioid receptor (MOR), κ-opioid receptor (KOR), δ-opioid receptor (DOR), and orphanin FQ/nociception (NOP) receptor.10,11 This opioid system is involved in a variety of regulatory functions including important roles in nociceptive, stress, emotional, and hedonic responses and modulation of thermoregulation, breathing, neuroendocrine function, GI motility, and immune responses. Various subtypes of opioid receptors have also been identified, with different pharmacologic functions. For example, at least three MOR subtypes have been described: μ1 is predominantly involved in opioid analgesia, μ2 is involved in opioid-induced respiratory depression, and μ3 is involved in opioid-induced immune suppression.12,13 Note, however, that the functional validation of most opioid receptor subtypes awaits the development of antagonists with sufficient selectivity to allow a clear differentiation by effect.
The endogenous opioid peptides include endorphins, enkephalins, and dynorphins, each of which display different affinities for the μ-, κ-, and δ-opioid receptors.14 β-endorphins have a high affinity for the MOR, met- and leu-enkephalins for the KOR, and Dynorphin A for the DOR. The recently discovered nociceptin has been identified as a selective endogenous ligand of the NOP  receptor,11,22 while endogenous morphine acts via the μ3-receptor located on immune cells, such as human monocytes.15
receptor,11,22 while endogenous morphine acts via the μ3-receptor located on immune cells, such as human monocytes.15
Opioids act not only through central and peripheral neuronal pathways, but also via non-neuronal mechanisms, such as actions on the immune system.16 Morphine and morphine-6-glucuronide (M6G), endogenously formed and stored in adrenal chromaffin cells and leukocytes, play an import role as modulators of non-neuronal responses in the immune system.17 Our considerations of endogenous opioid systems, therefore, have expanded from an opioid peptide system present in the central nervous system (CNS) and peripheral nervous system to include various peptide and non-peptide ligands in neuronal and non-neuronal cells throughout the body.
Opioid receptors are linked to G proteins in the cell membrane and are hence members of the large G-protein–coupled receptor (GPCR) family.18 GPCRs mediate a cascade of downstream signaling pathways leading to (i) the inhibition of adenyl cyclase and decreased cyclic AMP, (ii) activation of Ca2+ and K+ channels, and (iii) activation of MAPK/ERK, PKC, and P13 K/Akt.19 Interactions between opioid ligands and selective receptors have a number of important clinical effects. Morphine-induced analgesia and respiratory depression are both induced through activation of the MOR and subsequent activation of the adenylate cyclase/cyclic AMP pathway.20 Recently, opioid-induced activation of alternate cell signaling pathways through interactions with many (opioid and non-opioid) receptor types have been shown to affect the growth and distribution of cancer cells.19,21
Opioid Receptor Knockout Mice
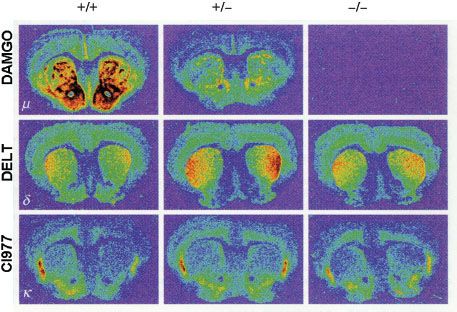
In the last decade a variety of mice lacking the various opioid receptors (“knockout mice”) have been bred to understand the molecular targets of exogenous opioids (Fig. 19-2).10 Inactivation of the μ-, κ-, and δ-opioid receptors, either separately or in combination, is not lethal. This suggests that the opioid receptor system is not crucial for development but is critical when experiencing stress or pain. For example, under conditions of fight or flight, recruitment of the endogenous opioid system will result in development of endogenous analgesia allowing the subject to respond to an acute body insult and consequently increases the chance of survival.
FIGURE 19-2. μ-, δ-, and κ-opioid receptor (MOR, DOR, and KOR, respectively) distribution in the brain of MOR knockout mice. The MOR is reduced in heterozygous MOR knockout mice and totally absent in homozygous knockout mice, while the other two receptor types remain present in both genotypes. +/+, wild-type mice; +/–, heterozygous MOR knockout mice; –/–, homozygous MOR knockout mice. Reprinted from: Matthes HWD, Maldonado R, Simonin F, et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the μ-opioid receptor gene. Science. 1996;383:819, with permission.
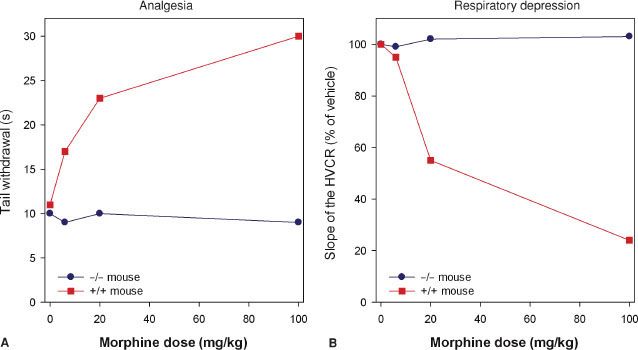
Studies indicate that morphine’s action at the MOR gene produces its clinical effects. Mice lacking the MOR gene do not experience morphine-induced analgesia, respiratory depression, reward and withdrawal, inhibition of GI transit, immunosuppression, or an increase in steroid hormones (Fig. 19-3).22–24 Analgesic responses after administration of other opioids, such as morphine’s metabolite M6G, are also absent in MOR knockout mice.25 These observations suggest the MOR is the target for both the desired and undesired effects of opioid analgesics and consequently designing a MOR activating drug that selectively produces desired effects such as analgesia, but not undesired effects such as life-threatening respiratory depression, is not possible.
FIGURE 19-3. Effect of morphine in mice lacking the μ-opioid receptor (–/–, homozygous μ-opioid receptor knockout mice) and mice with intact receptors (+/+, wild-type mice) on analgesic responses [(A) tail flick test in two mice] and respiratory responses [(B) the hypercapnic ventilatory response (HCVR) in two mice]. Data adapted from: Romberg.
Classification of Exogenous Opioids
Opioids may be classified on the basis of their synthesis, chemical structure, potency, receptor binding, and effect at the opioid receptors. According to their synthesis opioids are subdivided into natural (morphine), semisynthetic, and synthetic opioids. Natural opioid alkaloids are known as opiates, while the term opioids is used for all opioids (endogenous opioid peptides, natural opioid alkaloids, semisynthetic opioids, and synthetic opioids).
Semisynthetic opioids are derived from the morphine molecule and include buprenorphine, codeine, etorphine, heroin, hydromorphone, oxycodone, and oxymorphone. The synthetic opioids comprise the piperidines (e.g., loperamide, meperidine, alfentanil, fentanyl, sufentanil, remifentanil) and the methadones (e.g., methadone, dextro-propoxyphene).
 Opioid potency ranges from weak opioids such as codeine, dextro-propoxyphene, tramadol, and hydrocodone to strong opioids, which include etorphine, fentanyl, sufentanil, alfentanil, and remifentanil. Medium potency opioids include morphine, methadone, oxycodone, hydromorphone, and buprenorphine. Irrespective of the “strength” of these agents, all of these agents may potentially produce serious and potentially life-threatening side effects including sedation and respiratory depression, hypotension, and bradycardia. During surgery strong opioids are used in high doses while in the postoperative phase medium strength opioids such as morphine or methadone are used for treatment of acute pain. In 1986, the World Health Organization designed a stepwise approach for treatment of chronic cancer pain in which weak opioids are prescribed before strong opioids (http://www.who.int/cancer/palliative/painladder/en/).
Opioid potency ranges from weak opioids such as codeine, dextro-propoxyphene, tramadol, and hydrocodone to strong opioids, which include etorphine, fentanyl, sufentanil, alfentanil, and remifentanil. Medium potency opioids include morphine, methadone, oxycodone, hydromorphone, and buprenorphine. Irrespective of the “strength” of these agents, all of these agents may potentially produce serious and potentially life-threatening side effects including sedation and respiratory depression, hypotension, and bradycardia. During surgery strong opioids are used in high doses while in the postoperative phase medium strength opioids such as morphine or methadone are used for treatment of acute pain. In 1986, the World Health Organization designed a stepwise approach for treatment of chronic cancer pain in which weak opioids are prescribed before strong opioids (http://www.who.int/cancer/palliative/painladder/en/).
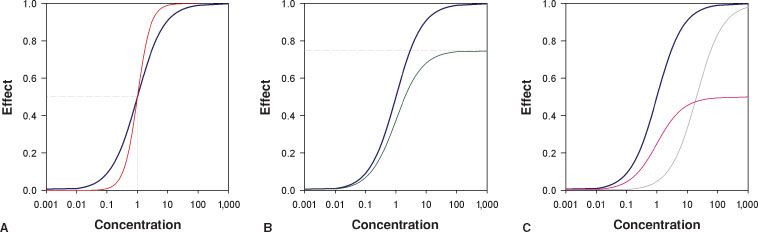
Opioids may be full agonists which cause the maximum possible effect when activating their receptors. Opioid full agonists at the MOR include morphine, piperidines, and methadone. Opioid partial agonists activate their receptor but cause only a partial or reduced effect (Fig. 19-4). Buprenorphine acts as a partial agonist at the MOR while it is a full agonist at the NOP and DOR.26 In clinical practice buprenorphine behaves as a full agonist with respect to pain relief yet its respiratory depressant effects are limited and display a “ceiling effect”.27 At the KOR buprenorphine acts as a competitive antagonist, which classifies this opioid also as a mixed agonist–antagonist. Naloxone and naltrexone are opioid antagonists.
FIGURE 19-4. Sigmoid Emax relationships for (A) full opioid agonists with C50 = 1 and steepness parameter γ = 1 (blue line) and γ = 2 (red line), (B) full opioid agonist (blue line with Emax = 1) and a partial agonist (green line with Emax = 0.75), (C) the rightward shift (gray line) observed by adding a competitive antagonist (such as naloxone) on top of a full agonist (blue line) causing an increase in C50 and the downward shift of adding a noncompetitive antagonist (purple line) to the full agonist causing an effect similar to that observed in (B) (i.e., a partial agonistic effect).
A more practical classification of opioids is their subdivision into agents with a rapid onset and offset of action (e.g., remifentanil and alfentanil) and agents with a slow onset/offset of action (e.g., morphine and buprenorphine). The concept of onset/offset of action will be discussed below.
Opioids Acting at Opioid and Non-opioid Receptors
Most opioid analgesics act at multiple receptor systems with different affinities. For example, morphine acts with high affinity at the MOR and with lower affinities at the KOR and DOR. Various opioids including methadone, dextro-propoxyphene and ketobemidone are opioid analgesics and also antagonists at the N-methyl-D-aspartate (NMDA) receptor.28,29 Of all opioids, methadone is the most potent NMDA receptor antagonist, being 6 to 18 times more potent than morphine in producing this effect. Antagonism of the NMDA receptor is clinically useful in reducing opioid tolerance and opioid-induced hyperalgesia (OIH) and in chronic pain states leading to pain hypersensitivity.30
Tramadol has both opioidergic and monoaminergic activity at dorsal horn spinal synapses of the nociceptive pathways.31 The analgesic effect of tramadol is due to enhancement of spinal inhibition of 5-hydroxytryptamine (5HT) and norepinephrine reuptake, causing the accumulation of 5HT and norepinephrine in the dorsal spinal horn.
The latest “novel” opioid recently registered worldwide is tapentadol, a potent analgesic with reduced GI and CNS side effects.8,9 Tapentadol has a dual mechanism of action. It is active at the MOR at spinal and supraspinal sites and as a norepinephrine reuptake inhibitor (NRI) in the spinal cord. The combination of these two mechanisms has been designated as the MOR–NRI concept. The affinity of tapentadol for the MOR is 50-fold lower than that of morphine. However, due to synergy between the two mechanisms of action, tapentadol produces potent analgesia and is useful in the treatment of moderate to severe acute and chronic pain. Tapentadol differs from tramadol due to its lack of serotonergic effects resulting in a lower incidence of nausea and vomiting. Tapentadol’s low affinity for the MOR may limit its undesirable side effects although its respiratory side effects have not yet been fully studied.
Finally, non-opioids may also act at opioid receptors. An important example is ketamine, which is an NMDA receptor antagonist with affinity for multiple receptor systems including the opioid receptors.32 Its anesthetic properties are related to its effect at the NMDA receptors while its analgesic effects are predominantly due to MOR activation.
OPIOID MECHANISMS
Mechanisms of Opioid Analgesia
Whereas nociception is the reception of signals in the CNS that have been triggered by noxious stimulation, pain is the subjective perception of that noxious stimulation. Opioids modify both nociception and the perception of a noxious stimulus (emotional coloring of pain).
Different types of peripheral sensory nociceptors, often free nerve endings, are stimulated by tissue damage and the resulting pain information is transmitted to the spinal cord by two types of small diameter peripheral afferent fibers: Slow conducting, unmyelinated C-fibers (which cause a dull burning pain) and faster, thinly myelinated Aδ fibers (which cause sharp, pricking pain). Both types of primary afferent fibers enter the dorsal horn of the spinal cord and terminate in its superficial layers (lamina I–II). Projection neurons from these laminae give rise to the ascending pathways of the spinothalamic tract. Thalamic nuclei receive the nociceptive inputs and pass the information to key brain pain reception sites such as the periaqueductal gray (PAG), amygdala, and somatosensory cortex. Activation of the MORs extensively located in these higher brain centers stimulates analgesia by activating descending inhibitory pathways from the PAG and rostroventral medulla (RVM) that inhibit nociceptive dorsal horn neuron firing in the spinal cord.33,34 Opioids also exert actions in the cortex and limbic systems affecting cholinergic systems that lead to changes in arousal and pain perception.35
μ-opioid-induced analgesia and descending inhibitory pathways may be activated not only by exogenous opioids, but also by activation of endogenous opioid systems. Direct electrical stimulation of the PAG and RVM induces analgesia that may be reversed by opioid antagonists.36 The electrically stimulated sites overlap with the opioid receptor sites and with opioid-containing interneurons, linking together the actions of exogenously applied analgesic stimuli and endogenous opioid systems. Three major examples of analgesia driven by the endogenous opioid system are (1) stress-induced analgesia37; (2) placebo-induced analgesia38; and (3) conditioning pain modulation (CPM).39
1. Stress-induced analgesia. The endogenous opioid system is activated under stressful conditions, as demonstrated by the delayed onset of pain by soldiers wounded in battle.40 The same higher brain centers bearing MORs are involved in the implementation of stress-induced analgesia.
2. Placebo-induced analgesia. The endogenous opioid system also mediates placebo-induced analgesia, a reduction of pain  resulting from an expectation of pain relief. Studies using fMRI and PET show activation of the endogenous opioid systems and MORs in the brains of subjects receiving placebo described as an analgesic.41–43 Placebo-induced analgesia may be used to enhance opioid-derived analgesia.
resulting from an expectation of pain relief. Studies using fMRI and PET show activation of the endogenous opioid systems and MORs in the brains of subjects receiving placebo described as an analgesic.41–43 Placebo-induced analgesia may be used to enhance opioid-derived analgesia.
3. CPM. Formerly known as diffuse noxious inhibitory control, it is a condition in which pain arising from a noxious stimulus applied to one part of the body is decreased by application of a second remote noxious stimulus.39,44,45 CPM is caused by the activation of descending inhibitory pathways.
Opioids may also act at spinal sites to induce analgesia. In the superficial laminae of the dorsal horn, local neuronal circuits process both ascending and descending pain pathways and are regulated by local endogenous opioid circuits.
Peripheral Opioid Analgesia
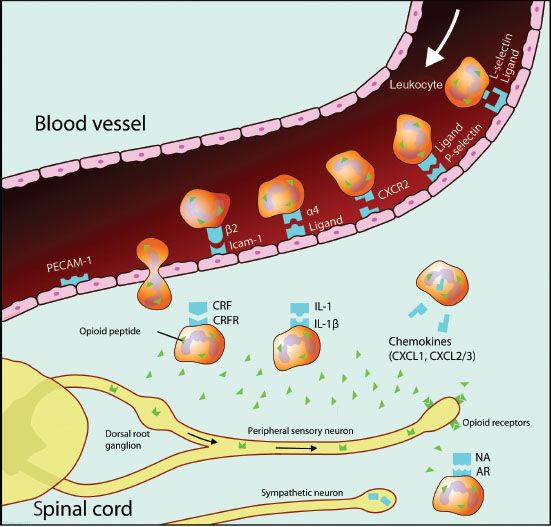
Opioids are also involved in peripheral analgesia by acting directly on sensory neurons (Aδ and C-fibers) to inhibit pain signal transmission. This is especially important in inflammatory pain. However, the immune system is also widely involved in peripheral analgesia.46 Opioid receptors are located not only on neurons, but on immune cells, such as human leukocytes.47 An insult to a peripheral tissue triggers the local release of many proinflammatory mediators that generate an inflammatory cascade, induce spontaneous nociceptor activity, and sensitize sensory neurons to induce spontaneous pain, allodynia (a non-painful stimulus is perceived as painful), and hyperalgesia (increased pain sensitivity). Early in the inflammatory process there is an influx of leukocytes into the inflamed area and these cells are a major source of opioid peptides to inflamed sites. Opioid peptides released locally interact with the opioid neuronal receptors to induce analgesia (Fig. 19-5).48 The inflammatory process also stimulates further opioid receptor upregulation and thereby increases the antinociceptive action of opioid peptides released by immune cells. In aggregate, the inflammatory process not only promotes inflammation and its painful sequelae, but also initiates and sustains a counteracting analgesia driven by endogenous opioids.49
FIGURE 19-5. Schematic diagram illustrating the role of opioids in analgesia of peripheral inflammation. Opioid-containing leukocytes are attracted to inflamed tissue by various chemokines and cytokines. Specific upregulated protein facilitates leukocyte migration through the vascular endothelium. In the inflamed tissue leukocytes interact with releasing agents such as corticotropin-releasing factor (CRF), interleukin-1 (IL-1), and norepinephrine (NA) derived from postganglionic sympathetic neurons, to secrete opioid peptides. These bind to peripheral opioid receptors, synthesized in the dorsal root ganglia and transported to peripheral endings of sensory neurons, to mediate analgesia. AR, adrenergic receptor; CRFR, corticotropin-releasing factor receptor; PECAM-1, platelet endothelial adhesion molecule 1; Icam-1, intracellular adhesion molecule 1; CXCR2, chemokine (C-X-C motif) receptor 2; CXCL1, chemokine (C-X-C motif) ligand 1; CXCL2/3, chemokine (C-X-C motif) ligand 1/2.
Opioid-induced Hyperalgesia (OIH) and Tolerance
Opioids can induce the paradoxical effect of OIH or an increase in pain sensitivity.50 OIH may limit the analgesic effects of opioids. During long-term and/or high-dose opioid treatment, rapid opioid dose escalation, or administration of an opioid with rapid onset/offset (e.g., remifentanil), a paradoxical increase in pain accompanies the treatment escalation.51,52 The MOR is not a prerequisite for OIH, as there is ample evidence from knockout mice  studies (devoid of MOR) or studies in mice treated with naloxone or naltrexone that OIH develops in response to exposure to high-dose opioids.53–55 There may be various mechanisms for OIH, including activation of the central glutaminergic system, central nitric oxide production, and facilitation of descending pronociceptive systems.
studies (devoid of MOR) or studies in mice treated with naloxone or naltrexone that OIH develops in response to exposure to high-dose opioids.53–55 There may be various mechanisms for OIH, including activation of the central glutaminergic system, central nitric oxide production, and facilitation of descending pronociceptive systems.
Postoperative patients who have received remifentanil infusions intraoperatively can have a higher incidence of OIH and need greater doses of morphine for control of postoperative pain than patients receiving non-remifentanil–based anesthesia.51 Although animal and human data indicate that all μ-opioids may cause OIH there seems to be a gradual difference in prevalence, with most observation of OIH following remifentanil treatment. This high incidence of OIH following remifentanil infusions may be related to its rapid offset of analgesia. In order to prevent severe pain responses following remifentanil-based anesthesia, administration of morphine (0.1 to 0.25 mg/kg) 45 to 60 minutes before the end of surgery is advisable, and adding a low-dose ketamine infusion may prevent the development of OIH (dose range: 10 to 30 mg/hr) due to ketamine’s NMDA antagonistic properties.52
OIH is not the same phenomenon as opioid tolerance.50 Acute opioid tolerance due to tachyphylaxis requires increasing doses of the opioid to reach a specific analgesic end point during the initial hours of opioid treatment. Chronic tolerance, often seen in opioid abusers, occurs over days and manifests as a decreasing analgesic effect, resulting in dose escalation and increasing the likelihood of OIH. In contrast to OIH, opioid receptor–related and post-activation intracellular processes play an important role in the development of tolerance (including β-arrestin–dependent receptor desensitization and internalization, and G-receptor uncoupling).56,57 Finally, pseudo-tolerance is a phenomenon seen in chronic pain patients due to progression of disease with an increase in the level of nociception often due to destruction of nerves in the tumor region, resulting in neuropathic pain, which is poorly responsive to opioid dose escalation.
ROUTES OF ADMINISTRATION
The most important and predictable route of administration of opioids perioperatively is the intravenous route, since the amount of drug entering the systemic circulation is precisely known. Non-intravenous administration routes (e.g., oral, subcutaneous, transdermal, inhalational, sublingual, oral transmucosal, intranasal, or rectal routes) are often more convenient for the patient although they come at the cost of more variable (and often slower) rates of absorption and bioavailability. Opioids given orally have bioavailabilities of 20% to 40% due to a rapid first-pass effect as a result of opioid metabolism in the liver.
Opioids given via epidural (e.g., patient-controlled epidural analgesia) and intrathecal routes need to diffuse into the surrounding nerve tissue and spinal cord to activate MORs. More lipophilic opioids (fentanyl, sufentanil) will penetrate faster and achieve higher concentrations into the spinal cord than hydrophilic opioids such as morphine and meperidine, yet they are also cleared from the spinal fluid more rapidly.58 Thus the lipophilic opioids may cause an early-onset respiratory depression whereas morphine tends to cause late ventilatory depression due to its slow clearance from the spinal fluid. After the epidural administration of opioids, lipophilic drugs are rapidly absorbed into the systemic circulation resulting in plasma concentrations similar to those observed after low-dose intravenous injections.58 The clinical effects of lipophilic opioids given epidurally are due to systemic, spinal, and supraspinal sites of action. It is important to note that epidurally administered opioids may exacerbate the risk of serious respiratory depression when coadministered with other parenteral opioids.
Intramuscular injections of opioids for treatment of postoperative pain should be avoided as there are superior alternatives such as the intravenous administration of opioids using patient-controlled analgesia (PCA) devices. Compared to IM injections intravenous PCA is associated with lower pain scores and greater patient satisfaction. PCA also does not require frequent painful injections. Under specific and exceptional circumstances, nurse-administered interval IM analgesia is acceptable, such as when the patient is unable to use the PCA device (mentally or physically incapacitated patients) or in case of fear of PCA by proxy.59 The latter is the case when family members press the PCA button.
PHARMACOKINETICS (PK) AND PHARMACODYNAMICS (PD)
When injected intravenously opioids are rapidly transported to the heart and pulmonary vessels from where they are dispersed to the various organs and tissues. After a standard dose of opioid, the inter-patient variability of effect is large and related to various factors including weight-related parameters (lean and fat body mass), organ function (hepatic and renal function), and cardiac output. This variability is manifest in the distribution and elimination constants that describe the PK profile of these drugs, which is also related to their physicochemical properties, such as molecule size, pKa (affects the degree of ionization of the molecule and depends on the plasma pH), protein binding (to albumin and α1-acid glycoprotein), and lipid solubility. These factors affect the passage of the drug into the brain across the blood–brain barrier and hence affect both the opioid’s PK and PD characteristics. For example, a small increase in pH seen with respiratory alkalosis will increase the non-ionized form of morphine, fentanyl, sufentanil, and remifentanil, which subsequently crosses the blood–brain barrier. Different drugs may also affect the blood–brain barrier active transport systems that eliminate opioids from the brain. For example, cyclosporine enhances morphine’s analgesic effect but not that of methadone, suggesting that cyclosporine selectively interferes with morphine’s efflux from the brain via specific transporter proteins.
When an opioid is injected into the venous system, there is an initial rapid peak in plasma concentration. Next, the drug rapidly enters multiple organ systems with high blood flow (such as the brain, liver, kidney) from which the plasma drug concentration rapidly drops followed by a slower drop due to redistribution to organs (such as the muscles and later tissues with high fat content) that are less well perfused. These concentration changes over time can be described by non-compartmental and compartmental PK models. Non-compartmental analysis describes the drug’s PK behavior in terms of volume of distribution (VD = drug dose/steady-state plasma drug concentration), rapid and slow distribution half-lives, and elimination half-life (t½elim). A high VD is observed for lipophilic opioids with low protein-binding affinity such as fentanyl (VD = 300 L) but a low VD is observed for remifentanil and alfentanil, due to a high clearance (remifentanil) and/or high protein binding. When VD is small, clearance is responsible for the drop in plasma concentration and consequently the loss of analgesia, whereas redistribution accounts for loss of analgesic effect in drugs with a high VD.
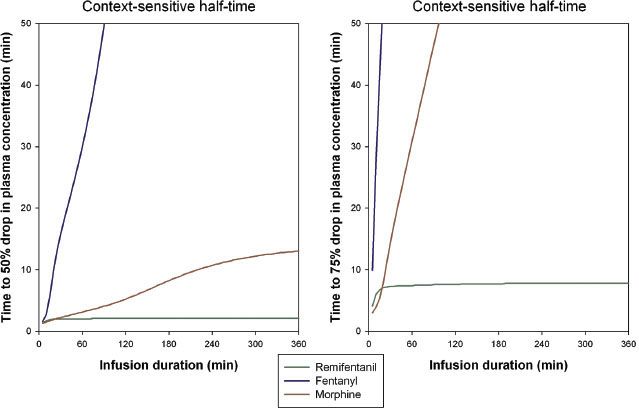
The time needed for the drug’s plasma concentration to decrease by 50%, from a steady-state plasma concentration after the drug infusion has stopped, is called the context-sensitive half-time  (CSt½) (Fig. 19-6).60,61 The CSt½ depends on the duration of the infusion, which is the context to which the term applies. For fentanyl, the context-sensitive half-time increases with the duration of the infusion, while for remifentanil the half-time is independent of the duration because of its rapid clearance (50% drop in plasma concentration is 2 minutes, 75% drop is 8 minutes).61 In clinical practice, the time to the loss of analgesia depends on the opioid dose, neuronal and receptor kinetic processes, the transport of the opioids from brain to plasma, and the context-sensitive half-time. The time course of a specific effect is difficult to predict for individual patients. For some side effects such as opioid-induced respiratory depression, the prediction of onset or offset of effect is even more complicated due to counteracting forces, such as the respiratory stimulant effects of increased arterial CO2 and the presence of pain.62
(CSt½) (Fig. 19-6).60,61 The CSt½ depends on the duration of the infusion, which is the context to which the term applies. For fentanyl, the context-sensitive half-time increases with the duration of the infusion, while for remifentanil the half-time is independent of the duration because of its rapid clearance (50% drop in plasma concentration is 2 minutes, 75% drop is 8 minutes).61 In clinical practice, the time to the loss of analgesia depends on the opioid dose, neuronal and receptor kinetic processes, the transport of the opioids from brain to plasma, and the context-sensitive half-time. The time course of a specific effect is difficult to predict for individual patients. For some side effects such as opioid-induced respiratory depression, the prediction of onset or offset of effect is even more complicated due to counteracting forces, such as the respiratory stimulant effects of increased arterial CO2 and the presence of pain.62
FIGURE 19-6. Context-sensitive half-times for remifentanil, fentanyl, and morphine. Left: Time to 50% drop in concentration versus infusion duration. Right: Time to 75% drop in concentration versus infusion duration.
Metabolism: Which Pathways and Metabolites are Clinically Relevant?
Most opioids are metabolized in the liver through either phase I (oxidative and reductive reactions catalyzed by the cytochrome P450 enzyme system) or phase II reactions (conjugation to a specific substrate). Metabolism may occur at other sites as well, such as in the enterocytes of the gastric tract, the kidney, or the brain. Excretion of the parent drug and/or metabolites occurs via the kidney and/or via the biliary tract into the gut where some opioids (morphine, buprenorphine) may undergo reuptake of the compound into the blood stream.
Three aspects of opioid metabolism have clinical importance:
1. Medications that inhibit or induce the CYP450 system may increase or decrease the clinical effect of opioids by interfering with their metabolism (Table 19-1).63,64
TABLE 19-1. INHIBITORS AND INDUCERS OF CYP3A AND INHIBITORS OF CYP2D6

 2. Opioid metabolites may either be active or inactive, which applies not only to their analgesic effect but also to their unwanted side effects.65,66
2. Opioid metabolites may either be active or inactive, which applies not only to their analgesic effect but also to their unwanted side effects.65,66
3. Genetic variability in the CYP system has clinical implications that are discussed in the section on Pharmacogenetics.
Morphine
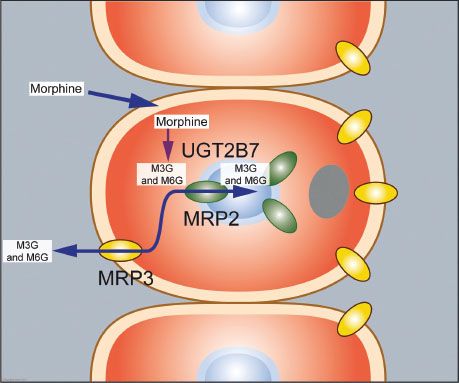
Morphine undergoes rapid metabolism (by UGT2B7, a phase II reaction) in the liver and within minutes after its administration the two most important hydrophilic metabolites appear in plasma: Morphine-3-glucuronide (M3G) and M6G.64–66 M3G is the major metabolite and about 60% of morphine is converted into M3G, while just 5% to 10% is converted to M6G. In humans M3G is without any analgesic or anti-analgesic action. M6G is a full MOR agonist but at the concentrations observed following morphine administration in a patient with normal renal function its contribution to the overall analgesic effect is minimal.67 Due to its low lipophilicity, passage of M6G across the blood–brain barrier is slow and consequently limited. In the hepatocytes both M3G and M6G are transported back into the bloodstream while a small part is transported into the bile ducts.68 In the gut both glucuronides are deglucuronidated and the resultant morphine molecule is partly absorbed by the enterocytes (Fig. 19-7). Enterocytes are able to metabolize morphine and transport the resultant M3G and M6G to the bloodstream (the enterohepatic cycle).
FIGURE 19-7. Morphine metabolism in the liver and transport of its metabolites into the bloodstream and bile system. Morphine enters the hepatocyte where it undergoes metabolism by UGT2B7 (a phase II reaction) into morphine-3-glucuronide (M3G) and morphine-6-glucuronide (M6G, not shown). These two glucuronides are transported via transporter proteins MRP3 (yellow) and MRP2 (green) back into the systemic circulation and into the biliary duct system.
Since the morphine-glucuronides are excreted via the kidney, patients with renal failure are at risk for M6G-related side effects.67 Since M6G is a full MOR agonist these side effects are typical of opioids and, most importantly, include sedation and respiratory depression. In patients with compromised renal function morphine treatment causes M6G to accumulate in high concentrations that may cause loss of consciousness and severe respiratory depression.67,69 Attempts to market M6G as a potent opioid analgesic have failed, likely due to its slow onset/offset time (t½ke0 4 to 6 hours) and low potency in humans.
Piperidines
Fentanyl, alfentanil, sufentanil, and remifentanil are lipophilic opioids that rapidly cross the blood–brain barrier. Fentanyl, alfentanil, and sufentanil are metabolized by the liver, catalyzed by the cytochrome P450 enzyme system.64,70,71 Fentanyl has a high hepatic extraction ratio with clearance approaching liver blood flow (1.5 L/min). The major metabolite of fentanyl is the inactive compound norfentanyl. Sufentanil also has a high hepatic extraction ratio with a clearance of 0.9 L/min. Alfentanil is metabolized by CYP3A4 and 3A5 forming the inactive compounds noralfentanil and N-phenylpropionamide. The polymorphic expression of the CYP3A5 gene accounts for the great variability in alfentanil metabolism and clearance.72 Remifentanil contrasts with the other piperidines in that it is not metabolized in the liver.73 Remifentanil contains a methyl ester side chain (Fig. 19-1) that is metabolized by blood (within the erythrocyte) and tissue nonspecific esterases. This causes a rapid clearance of the drug (context sensitive half-life of 2 minutes) making it the most rapidly acting opioid currently available. Clearance of remifentanil is 3 to 5 L/min, which exceeds liver blood flow affirming its extrahepatic clearance. Remifentanil is usually administered as a continuous infusion since its plasma level decreases by 50% in as little as 40 seconds.74–76
Methadone
Methadone is extensively metabolized to an inactive form by CYP2B6, which is also affected by pharmacogenetic variability.77,78 Methadone has a 60% to 95% bioavailability, high potency, and a long duration of action. Furthermore, there is considerable variation among recipients in the response to the drug. While methadone has properties which make it attractive for use intravenously as a perioperative analgesic, in a controlled and well-monitored environment, these same properties may prove hazardous when methadone is administered orally for treatment of patients with chronic pain. Large numbers of patient deaths have been attributed to the long, and often unpredictable, duration of action of methadone when administered orally.
Naloxone
Naloxone is the most valuable and popular nonspecific MOR antagonist.79 Since it has a low and unpredictable bioavailability after oral intake due to an extensive (>95%) first-pass effect, naloxone is best given via the intravenous route.79 The most important metabolic pathway of naloxone is glucuronidation into the inactive naloxone-3-glucuronide. Its duration of effect is short, ranging from 15 to 45 minutes, which requires it to be redosed or administered as a continuous infusion when antagonism is required for long-acting opioids or for patients experiencing an opioid overdose.
PKPD Models of Opioid Effect: Which End Point Serves the Clinician Best?
 The PK of a drug describes the time course of dose to concentration; the PD describes the concentration to effect relationship;
The PK of a drug describes the time course of dose to concentration; the PD describes the concentration to effect relationship;  the effect can be any of the desired or undesired drug effects. Pharmacokinetic–pharmacodynamic (PKPD) models are constructed for each drug to allow the clinician to understand and predict the clinical implication of a given dose to a desired effect.80 These models allow dosing regimens to be constructed on the basis of patient characteristics such as total or lean body weight, gender, age, and other characteristics, making them particularly helpful when treating individual patients. The PK part of such models describes the drug distribution kinetics. This relates to both the parent drug and the possible metabolites. In compartmental models, the concentration–time profiles are described by drug transfer between interconnected hypothetical compartments, mimicking drug absorption, distribution, elimination, and metabolism. The PD part of the model describes the drug concentration–effect relationship. This hypothetical effect compartment is made infinitely small so that it does not influence the drug’s disposition (PK) and is located at the drug’s target organ, such as the muscle endplate for muscle relaxants and the brain for hypnotic drugs. For most opioid effects (such as analgesia, sedation, and respiratory depression) the effect site is located within the CNS while the effect site for constipation is the GI tract. The delay between the peak drug concentration in the plasma and the peak concentration at the effect site is described by the plasma–effect-site
the effect can be any of the desired or undesired drug effects. Pharmacokinetic–pharmacodynamic (PKPD) models are constructed for each drug to allow the clinician to understand and predict the clinical implication of a given dose to a desired effect.80 These models allow dosing regimens to be constructed on the basis of patient characteristics such as total or lean body weight, gender, age, and other characteristics, making them particularly helpful when treating individual patients. The PK part of such models describes the drug distribution kinetics. This relates to both the parent drug and the possible metabolites. In compartmental models, the concentration–time profiles are described by drug transfer between interconnected hypothetical compartments, mimicking drug absorption, distribution, elimination, and metabolism. The PD part of the model describes the drug concentration–effect relationship. This hypothetical effect compartment is made infinitely small so that it does not influence the drug’s disposition (PK) and is located at the drug’s target organ, such as the muscle endplate for muscle relaxants and the brain for hypnotic drugs. For most opioid effects (such as analgesia, sedation, and respiratory depression) the effect site is located within the CNS while the effect site for constipation is the GI tract. The delay between the peak drug concentration in the plasma and the peak concentration at the effect site is described by the plasma–effect-site  equilibration constant ke0 (or its half-life t½ke0 = ln 2/ke0),81–83 which is commonly referred to as hysteresis. For the analgesic and respiratory depressive effects of opioids, the hysteresis is determined by the drug’s passage across the blood–brain barrier (the more lipophilic an opioid, the faster the transfer into the brain compartment), receptor kinetics, and neuronal dynamics. The effect-site concentration–effect relationship is described by a sigmoid Emax model80–84: Effect = (CE/C50)γ/[1 + (CE/C50)γ], where CE is the drug concentration in the hypothetical effect site, C50 is the measure of drug potency or the effect-site or steady-state concentration causing 50% of the effect, and γ is the Hill or steepness parameter (Fig. 19-4). In summary, any PKPD analysis using the above-mentioned descriptions yields PK parameters (volumes of distribution and clearances), as well PD parameters related to drug potency (C50) and the onset/offset times of the drug (t½ke0). It is important to understand that PK (volumes of distribution and rate constant) and PD (potency) values vary largely among patients. This is related to differences in physiology, underlying disease, age, weight, ethnicity, and other factors. Thus the clinician should choose a PK/PD set derived from a population of subjects whose characteristics are most similar to the individual they are treating. For example, due to changes in PK and PD behavior, elderly patients display a greater opioid sensitivity85; patients with liver or renal insufficiency require adaptation to their dosing; and patients with certain genetic abnormalities may experience unusual responses to opioids (see later).63,64
equilibration constant ke0 (or its half-life t½ke0 = ln 2/ke0),81–83 which is commonly referred to as hysteresis. For the analgesic and respiratory depressive effects of opioids, the hysteresis is determined by the drug’s passage across the blood–brain barrier (the more lipophilic an opioid, the faster the transfer into the brain compartment), receptor kinetics, and neuronal dynamics. The effect-site concentration–effect relationship is described by a sigmoid Emax model80–84: Effect = (CE/C50)γ/[1 + (CE/C50)γ], where CE is the drug concentration in the hypothetical effect site, C50 is the measure of drug potency or the effect-site or steady-state concentration causing 50% of the effect, and γ is the Hill or steepness parameter (Fig. 19-4). In summary, any PKPD analysis using the above-mentioned descriptions yields PK parameters (volumes of distribution and clearances), as well PD parameters related to drug potency (C50) and the onset/offset times of the drug (t½ke0). It is important to understand that PK (volumes of distribution and rate constant) and PD (potency) values vary largely among patients. This is related to differences in physiology, underlying disease, age, weight, ethnicity, and other factors. Thus the clinician should choose a PK/PD set derived from a population of subjects whose characteristics are most similar to the individual they are treating. For example, due to changes in PK and PD behavior, elderly patients display a greater opioid sensitivity85; patients with liver or renal insufficiency require adaptation to their dosing; and patients with certain genetic abnormalities may experience unusual responses to opioids (see later).63,64
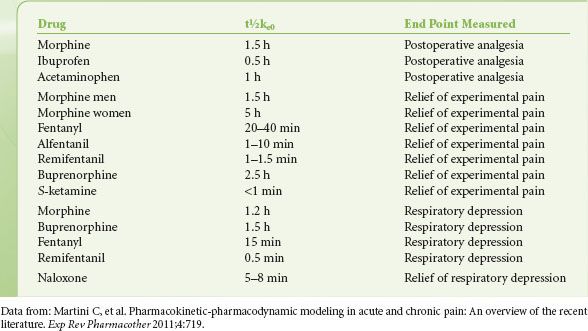
For most opioids, the target effect when constructing PKPD models has traditionally been the slowing of the frequency components of the EEG, quantified by a shift in the 95th percentile of the power spectrum (95% spectral edge frequency).86,87 The C50 and t½ke0 derived from these studies are useful to compare the potency and onset/offset of opioids. However, since the C50 for EEG effects occurs beyond the normal clinical dose range of opioids, more clinically useful C50 values would include those for the analgesic, respiratory depressive, and sedative effect of opioids. For alfentanil and fentanyl C50 values range from 75 and 1 ng/mL for sedation to 150 and 2 ng/mL for analgesia, respectively.88,89 This indicates that these clinically relevant effects occur at lower doses than their effects on the EEG. For fentanyl it is of further interest that t½ke0 values for analgesia (20 to 40 minutes) and respiratory depression are much longer (15 minutes) than those observed for EEG-slowing (5 to 6 minutes). In Table 19-2 values of t½ke0 for the end points of pain relief and respiratory depression are given for various analgesics currently in use.
TABLE 19-2. ESTIMATES OF THE T½KE0 FOR A VARIETY OF ANALGESICS DETERMINED FOR CLINICALLY RELEVANT END POINTS OF PAIN RELIEF AND RESPIRATORY DEPRESSION
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


