FIGURE 16.1 Illustration showing the pathogenesis of acute coronary syndromes. Rupture of an atherosclerotic plaque leads to activation of platelets and clotting factors (upper panel), and results in the formation of an occlusive thrombus (lower panel).
Thrombocentric Management
The discovery that coronary thrombosis is the culprit in acute coronary syndromes has resulted in a number of therapeutic measures aimed at alleviating the thrombotic obstruction and preventing recurrences. These measures include the following:
1. Therapy with fibrinolytic drugs to promote clot dissolution.
2. Balloon catheter angioplasty to restore patency in obstructed coronary arteries, and vascular stenting to maintain patency.
3. Antiplatelet therapy (with aspirin, clopidogrel, and glycoprotein IIb/IIIa inhibitors) and anticoagulation therapy (with heparin) to prevent reocclusion in infarct-related arteries after flow is restored by thrombolysis or angioplasty.
Each of these measures is described in this chapter, along with other measures designed to prevent unwanted cardiac stimulation in the setting of compromised coronary blood flow.
ROUTINE MEASURES
The following measures are used when ACS is first suspected, often before the diagnostic evaluation is completed. Some of these interventions, which are summarized in Table 16.1, are used in the pre-hospital setting.
Table 16.1 Routine Measures in Acute Coronary Syndromes

Oxygen Therapy
Providing supplemental O2 has been a standard practice in patients with ACS (3,5), even when arterial oxygenation is normal. This practice has been questioned in recent years (12) because oxygen promotes coronary vasoconstriction (oxygen is a vasoconstrictor in all organs except the lungs, where it acts as a vasodilator), and because oxygen is the source of toxic oxygen metabolites (see Figure 14.1), which have been implicated in reperfusion injury (13). The deleterious effects of oxygen are emphasized in a recent study showing an increased risk of unfavorable outcomes associated with O2 therapy in acute MI (14).
Concern about the potential for harm from unregulated O2 breathing is evident in the most recent guidelines on acute coronary syndromes from the American Heart Association (2), which recommends supplemental O2 only when the arterial O2 saturation (SaO2) falls below 94% (2). This is higher than the standard threshold for O2 breathing (SaO2 <90%), but it is still a significant step in the realization that oxygen can be a toxic gas.
Relieving Chest Pain
Relieving chest pain not only promotes a sense of well-being, it also helps to alleviate unwanted cardiac stimulation from anxiety-induced adrenergic hyperactivity.
Nitroglycerin
Nitroglycerin (0.4 mg) is given as a sublingual tablet or aerosol spray to relieve chest pain, and a total of 3 doses can be given in 5-minute intervals if necessary. (The mechanism of pain relief with nitroglycerin is un-clear. It has been attributed to the coronary vasodilator effects of nitroglycerin, but other coronary vasodilators, such as, nitroprusside, do not relieve ischemic chest pain.) If the pain subsides, an intravenous infusion of nitroglycerin can be started for continued pain relief using the dosing regimen in Table 16.1. Intravenous nitroglycerin can also be used as a systemic vasodilator when ACS is accompanied by hypertension or decompensated heart failure (2). Chest pain that is not relieved by nitroglycerin should prompt the immediate administration of morphine.
CAVEATS: Nitroglycerin is not advised when right ventricular infarction is suspected (because the venodilator effects of nitroglycerin are counterproductive in this condition), and in patients who have taken a phosphodiesterase inhibitor for erectile dysfunction within the past 24 hours (because of the risk of hypotension) (3,5,8).
Morphine
Morphine is the drug of choice for chest pain that is refractory to nitroglycerin. The initial dose is 2 to 4 mg by slow intravenous push, and this can be followed by repeat doses of 2 to 8 mg every 5 or 10 minutes if needed (3).
Morphine administration is often followed by a modest decrease in heart rate and blood pressure, which is an expression of the decreased adrenergic system activity that accompanies pain relief. A drop in blood pressure to hypotensive levels often indicates hypovolemia, and can be corrected with fluid challenges (5). Morphine occasionally produces bradycardia and hypotension (vagomimetic effect), which can be managed with atropine (0.5–1.5 mg IV) if necessary (3). Morphine-induced respiratory depression is uncommon in ACS (3,5).
Aspirin
Aspirin is a well-known antiplatelet agent that produces an irreversible inhibition of platelet aggregation by inhibiting thromboxane production (15). When started within 24 hours of symptom onset in ACS, aspirin has been shown to reduce the mortality rate (absolute decrease is 2 to 3%) and the rate of re-infarction (16,17). As a result, aspirin is recommended for all patients with suspected ACS or a history of ACS (2–6).
In patients who are not taking aspirin regularly, the first dose of aspirin should be given as soon as possible after ACS is suspected. The initial dose is 162–325 mg, which should be given as chewable aspirin to enhance buccal absorption. This is followed by a daily aspirin dose of 75–162 mg, given as enteric-coated tablets. For patients with an aspirin allergy, clopidogrel (Plavix) is a suitable alternative (2–6). The dosing regimens for clopidogrel are presented later in the chapter.
β-Receptor Antagonists
The benefit of β-Receptor antagonists in ACS is based on their ability to reduce cardiac work and decrease myocardial energy requirements. Early institution of beta-blocker therapy is recommended for all patients with ACS who do not have a contraindication to β-Receptor blockade (2,3,5,7,8). These contraindications include high-grade AV block, systolic heart failure, hypotension, and reactive airway disease. In addition, β-Receptor antagonists should not be used for cocaine-induced chest pain or myocardial infarction because there is a risk of aggravated coronary vasospasm from unopposed α-adrenergic receptor activity (3).
Oral β-blocker therapy is suitable for most cases of ACS, while intravenous therapy is reserved for patients with persistent chest pain, tachycardia, and hypertension (2,3,7). The β-blockers used most often in clinical trials of ACS are atenolol (Tenormin) and metoprolol (Lopressor), and the dosing regimens for these drugs is shown in Table 16.1.
REPERFUSION THERAPY
The principal determinant of outcomes in acute coronary syndromes is the ability to restore patency in occluded coronary arteries using thrombolytic therapy or percutaneous coronary intervention (PCI), which includes coronary angioplasty and stent placement, when indicated.
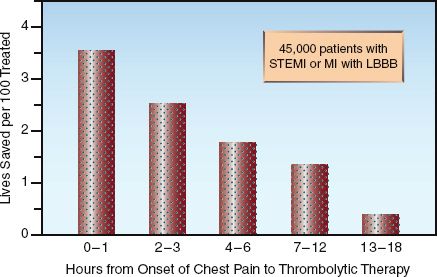
FIGURE 16.2 The survival benefit of thrombolytic therapy in relation to the time elapsed from the onset of chest pain to the start of therapy. STEMI = ST-segment elevation myocardial infarction, LBB = left bundle branch block. Data from Reference 19.
Thrombolytic Therapy
The evaluation of drugs that stimulate fibrinolysis began immediately after the discovery (in 1980) that transmural myocardial infarction was the result of occlusive coronary thrombosis. In 1986, the first clinical trial of thrombolytic therapy was published, and showed a time-dependent survival benefit in patients with ST-elevation MI (STEMI) (18).
Summary of Benefits
The following statements summarize the clinical experience with thrombolytic therapy in acute coronary syndromes.
1. Thrombolytic therapy provides a survival benefit in the following conditions (2,3,8):
a. Acute MI associated with ST-segment elevation of at least 0.1 mV or 1 mm in two contiguous leads (STEMI).
b. Acute MI associated with a new left bundle branch block.
c. Acute, posterior-wall MI, which is characterized by ST-depression in the anterior precordial leads and ST-elevation in extreme lateral precordial leads V7–V9 (20).
3. The survival benefit of thrombolytic therapy is time-dependent; i.e., it is greatest in the first few hours after the onset of chest pain, and is lost if more than 12 hours have elapsed from the onset of chest pain (2,3,8,19). This is demonstrated in Figure 16.2. Note the steady de-cline in survival benefit over the first 12 hours after the onset of chest pain. This data highlights the most important feature of reperfusion therapy: i.e., time lost is lives lost.
4. To ensure optimal benefit from thrombolytic therapy, the American Heart Association recommends that thrombolytic therapy should be started within 30 minutes after the initial presentation (2,3). Since most presentations occur in the emergency department, this is known as the door-to-needle time.
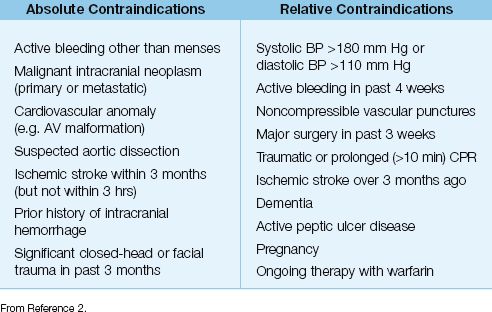
5. In addition to time constraints, the use of thrombolytic therapy is restricted by a number of contraindications, which are listed in Table 16.2.
Table 16.2 Contraindications to Thrombolytic Therapy
Thrombolytic Agents
Thrombolytic agents act by converting plasminogen to plasmin, which then breaks fibrin strands into smaller subunits. The fibrinolytic drugs used in ACS are shown in Table 16.3 These drugs act primarily on the plasminogen that is bound to fibrin (clot-specific fibrinolysis), and this limits the extent of systemic fibrinolysis and thereby limits the risk of unwanted hemorrhage.
ALTEPLASE (Activase) is a recombinant tissue plasminogen activator (tPA) that was popularized in 1993 by the GUSTO trial (21), which showed superior clinical outcomes with alteplase compared to streptokinase (the original fibrinolytic agent used in ACS). Alteplase is given as a fixed IV bolus dose followed by a 90-minute weight-based drug infusion (see Table 16.3). It has largely been replaced by the more rapid-acting fibrinolyitc agents, but there is no evidence of inferior clinical outcomes with alteplase compared to the more rapidly-acting fibrinolyitc agents (see next).
RETEPLASE (Retavase) is a recombinant variant of tPA that is given as an IV bolus (10 Units), which is repeated in 30 minutes. It produces more rapid clot lysis than alteplase (22), but clinical trials have shown no survival advantage with reteplase when compared to alteplase (23). Rete-plase is the only fibrinolytic agent that is given in fixed (not weight-based) doses, and this has created some popularity for the drug.
TENECTEPLASE (TNK-ase) is another variant of tPA that is given as a single IV bolus using a weight-based dosing regimen (see Table 16.3). It produces more rapid clot lysis than reteplase (24), but clinical trials have shown no survival advantage with tenecteplase compared to alteplase (25).
Table 16.3 Thrombolytic Agents and Dosing Regimens for MI

Major Bleeding
Clot-specific fibrinolytic agents produce some degree of systemic fibrinolysis, which can deplete circulating fibrinogen and create an increased risk of bleeding. The risk of major bleeding such as intracerebral hemorrhage (0.5–1%) and extracranial bleeding that requires blood transfusions (5–15%) is equivalent with alteplase, reteplase, and tenecteplase (24,26). Major bleeding from thrombolysis can be treated with cryoprecipitate (10 to 15 bags) followed by fresh frozen plasma (up to 6 units) if necessary (the goal is a serum fibrinogen ≥1 mg/ml). The use of antifibrinolytic agents such as epsilon-aminocaproic acid (5 grams IV over 15–30 min) is discouraged because of the risk for thrombosis (26).
Summary
Despite differences in pharmacokinetic properties, the fibrinolytic agents in Table 16.3 are equivalent in terms of survival benefit and risk of bleeding. The clinical experience with thrombolytic therapy then leads to the following summation: the important issue in thrombolytic therapy is not which agent to use, but how quickly to use it.
Percutaneous Coronary Intervention
In 1977, a Swiss cardiologist named Andreas Gruntzig used a homemade balloon-tipped catheter to reopen an occluded left main coronary artery. This “Gruntzig procedure” (coronary angioplasty) was promptly sanctioned by the American Heart Association, and was introduced in the 1980s as an alternative to thrombolytic therapy. (Unfortunately, Dr. Gruntzig died in a plane crash in 1985, just as his procedure was gaining widespread use.) In the late 1990s, the placement of stents was introduced to maintain vascular patency after coronary angioplasty. The combination of coronary arteriography, angioplasty, and stent placement is known as percutaneous coronary intervention (PCI).
PCI in STEMI
Several clinical trials have shown that PCI is superior to thrombolytic therapy for restoring flow in occluded arteries and reducing the incidence of unfavorable outcomes (1–3, 27–29). This is shown in Figure 16.3. The bar graphs on the left (depicting vascular events) show superior results with PCI for restoring normal flow in infarct-related arteries and preventing reocclusion. The bar graphs on the right (depicting clinical outcomes) show superior results with PCI for reducing the mortality rate and the reinfarction rate.
TIMING: The survival benefit of PCI is time-dependent, similar to thrombolytic therapy (see Figure 16.2). This is demonstrated in Figure 16.4, which shows the mortality rate (at 30 days) in relation to the time elapsed from hospital arrival to coronary angioplasty (the “door-to-balloon” time) (30). The mortality rate rises steadily with increasing delays to angioplasty, and the increased mortality becomes significant when the delay exceeds 2 hours. This is the basis for the recommendation that PCI should be performed within 90 minutes after hospital arrival (2–4).
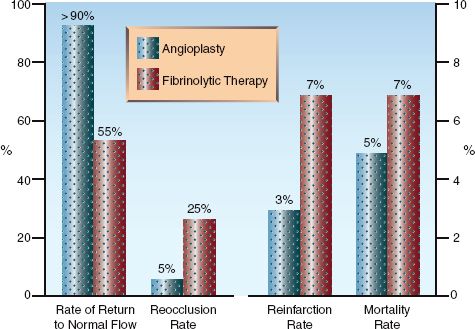
FIGURE 16.3 Comparative effects of coronary angioplasty and thrombolytic therapy on vascular events (graph on the left) and clinical outcomes (graph on the right) in patients with ST-elevation myocardial infarction. Data from references 27–29.
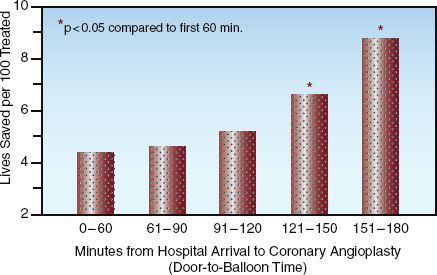
FIGURE 16.4 Mortality rate in relation to the time elapsed from hospital arrival to coronary angioplasty (door-to-balloon time). Asterisk indicates a significant difference compared to the initial time period (0–60 min). Adapted from data in Reference 30.
INTERHOSPITAL TRANSFER: The major limitation of PCI is availability; i.e., less than 25% of hospitals in the United States can provide PCI on a timely basis. One solution for this problem is a facilitated transfer from “have-not” hospitals to hospitals that can provide emergent PCI. Clinical studies have shown that interhospital transfer for PCI can result in a survival benefit if the transfer can be completed within 1–2 hours of the patient’s arrival at the “have not” hospital (31). Therefore, for patients who are candidates for reperfusion therapy, interhospital transfer for PCI is encouraged if the total door-to-balloon time, including the transfer time, does not exceed 90 minutes (2–4). Strict adherence to this time requirement is not inviolate because delayed PCI may still be a better option than no PCI (e.g., in patients with heart failure or hemodynamic instability).
Approach to Reperfusion Therapy in STEMI
Decisions regarding immediate reperfusion therapy in suspected cases of STEMI can be organized as a series of four questions, as shown in Table 16.4. The first two questions determine if the patient is a candidate for early reperfusion therapy, and the last two questions determine the type of reperfusion therapy that is most appropriate (i.e., PCI or thrombolytic therapy).
Other Indications for PCI
PCI is also advantageous in the following situations:
1. For patients with non-ST elevation MI (NSTEMI) who have risk factors for a poor outcome (e.g., unrelenting chest pain, heart failure, or hemodynamic instability) (4–6).
2. When thrombolytic therapy fails.
Table 16.4 Approach to Reperfusion Therapy in ST-Elevation MI

ADJUNCTIVE ANTITHROMBOTIC THERAPY
Antithrombotic therapy with heparin and antiplatelet agents other than aspirin have proven benefits in ACS when used with or without reperfusion therapy.
Heparin
Anticoagulation with heparin is beneficial in most patients with ACS. It is particularly advantageous after thrombolytic therapy, to counteract the prothrombotic actions of thrombin released by clot dissolution. The following is a summary of the recommendations for unfractionated heparin (UFH) and low-molecular-weight heparin (LMWH) in acute coronary syndromes. (See Chapter 6 for a description of the differences between UFH and LMWH.)
1. Short-term therapy with UFH is preferred for patients who receive reperfusion therapy with fibrinolytic agents or PCI. The recommended dosing regimens are as follows:
a. UFH with PCI: Use intravenous (IV) bolus doses of 70–100 Units/kg to maintain an activated clotting time of 250–350 seconds during the procedure (3). Reduce dose to 50–70 Units/kg if UFH is combined with glycoprotein receptor antagonists (see later) (7).
b. UFH with Thrombolytic Therapy: Start with an IV bolus dose of 60 Units/kg, and follow with an infusion of 12 Units/kg/hr for at least 48 hours (3). Adjust dosing to maintain an activated PTT (aPTT) at 1.5–2 times control. For a body weight >70 kg, the maximum doses are 4,000 Units (IV bolus) and 1,000 Units hourly (3).
2. LMWH is preferred for patients who do not receive reperfusion therapy. The dosing regimen for a popular LMWH (enoxaparin) is as follows:
a. Enoxaparin in ACS: 30 mg IV bolus, then 1 mg/kg by subcutaneous injection every 12 hours for the duration of hospitalization (7).
b. Enoxaparin in Renal Insufficiency: When creatinine clearance <30 mL/min, reduce the daily dose by 50% (e.g., 1 mg/kg every 24 hours) (7).
3. For patients with a history of heparin-induced thrombocytopenia (see Chapter 19), the following alternative regimens are available:
a. For PCI: Bivalirudin (a direct thrombin inhibitor), 0.7 mg/kg as IV bolus, followed by an infusion of 1.75 mg/kg/hr (7). Discontinue after successful PCI.
b. For Thrombolytic Therapy or No Reperfusion Therapy: Fondaparinux (factor Xa inhibitor), 2.5 mg by subcutaneous injection daily (7,8).
Thienopyridines
The thienopyridines (clopidogrel, ticlodipine, tigrecalor, prasugrel) are antiplatelet agents that irreversibly block surface receptors involved in ADP-induced platelet aggregation (32). This mechanism of action differs from that of aspirin, so these drugs can be used in addition to aspirin or as an alternative to aspirin. The thienopyridines are prodrugs that require activation in the liver, and are ineffective in patients with liver failure.
Clopidogrel
Clopidogrel (Plavix) is the most popular thienopyridine, and has a proven survival benefit when combined with aspirin in both STEMI and NSTEMI patients, with or without reperfusion therapy (1–6,32,33). Clopidogrel is also recommended as a replacement for aspirin in aspirin-allergic patients (3,5). The dosing recommendations for clopidogrel are as follows:
1. Clopidogrel without PCI: Start with an oral loading dose of 300 mg, given as soon as possible, and follow with an oral maintenance dose of 75 mg daily. The same dose is used when clopidogrel is added to aspirin or used as a replacement for aspirin.
2. Clopidogrel with PCI: Use an oral loading dose of 600 mg prior to PCI (4,6), and follow with the standard maintenance dose of 75 mg daily.
3. Clopidogrel and Surgery: Clopidogrel must be discontinued at least 5 days before major surgery is performed (4,6), so it is wise to avoid the drug if emergency coronary bypass surgery is anticipated.
The activation of clopidogrel in the liver is blocked by protein pump inhibitors (5,6,32). The clinical significance of this interaction is unclear, but it seems wise to avoid proton pump inhibitors for stress ulcer prophylaxis in patients receiving clopidogrel.
Glycoprotein Receptor Antagonists
When platelets are activated, specialized glycoprotein receptors on the platelet surface (named IIb and IIIa) change their configuration and begin to bind fibrinogen. This allows fibrinogen molecules to form bridges between adjacent platelets, which promotes platelet aggregation. Glycoprotein receptor antagonists (also called IIb/IIIa inhibitors) block fibrinogen binding to activated platelets and inhibit platelet aggregation. These drugs are the most potent antiplatelet agents available, and are sometimes referred to as the superaspirins.
Table 16.5 Antiplatelet Therapy with Glycoprotein Receptor Antagonists
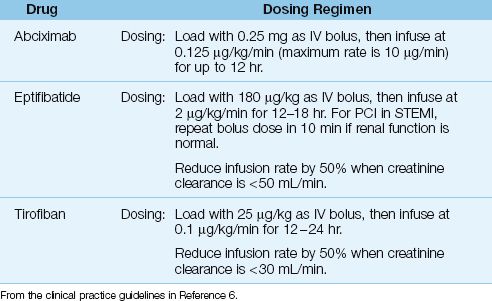
Drugs and Dosing Regimens
The IIb/IIIa inhibitors available for clinical use include abciximab (ReoPro), eptifibatide (Integrilin), and tirofiban (Aggrastat). All three are given by in-travenous infusion using the dosing regimens in Table 16.5. These drugs are used in high-risk patients who receive emergent PCI, and are given just before or at the start of the procedure (6,32). They are managed primarily by invasive cardiologists, and are described only briefly here.
Abciximab is (an almost unpronounceable name for) a monoclonal antibody that is the most potent, most expensive, and longest-acting IIb/IIa inhibitor. After discontinuing abciximab, bleeding times can take 12 hours to normalize (32). Eptifibatide (a synthetic peptide) and tirofiban (a tyrosine derivative) are short-acting agents that are cleared by the kidneys. After discontinuing these drugs, bleeding times return to normal in 15 minutes for eptifibatide and 4 hours for tirofiban (32). Dose adjustments are recommended for both drugs in renal insufficiency, as indicated in Table 16.5.
COMPLICATIONS
The appearance of decompensated heart failure and cardiogenic shock in the first few days after an acute MI is an ominous sign, and usually indicates a catastrophic structural defect like acute mitral regurgitation, or extensive muscle injury resulting in cardiac pump failure. Fatal outcomes are common in these conditions despite timely interventions.
Structural Defects
The following defects are usually the result of transmural (ST-elevation) infarctions.
Acute Mitral Regurgitation
Acute mitral regurgitation is the result of papillary muscle rupture, and presents with the sudden onset of pulmonary edema and the characteristic holosystolic murmur radiating to the axilla. The pulmonary artery occlusion pressure should show prominent V waves, but this can be a non-specific finding. Diagnosis is by echocardiography, and arterial vasodilators (e.g., hydralazine) are used to relieve pulmonary edema pending surgery. Mortality is 70% without surgery and 40% with surgery (34).
Rupture of Ventricular Septum
Rupture of the septum separating the two ventricles can occur anytime in the first 5 days after an acute MI, and the diagnosis can be elusive without cardiac ultrasound. There is a step-up in O2 saturation from right atrial to pulmonary artery blood, but this is rarely measured. Initial management involves vasodilator (e.g., nitroglycerin) infusions and the intra-aortic balloon pump if needed. Mortality is 90% without surgery and 20% to 50% with surgery (3).
Rupture of Ventricular Wall
Ventricular free wall rupture occurs in up to 6% of cases of STEMI, and is more common with anterior MI, fibrinolytic or steroid therapy, and advanced age (3). The first signs of trouble are usually return of chest pains and new ST-segment abnormalities on the ECG. Accumulation of blood in the pericardium often leads to rapid deterioration and cardiovascular collapse from pericardial tamponade. Diagnosis is made by cardiac ultrasound (if time permits), and prompt pericardiocentesis combined with aggressive volume resuscitation is required for hemodynamic support. Immediate surgery is the only course of action, but fewer than half of the patients survive despite surgery (3).
Cardiac Pump Failure
About 10% of cases of ST-elevation MI (STEMI) result in enough muscle damage to produce decompensated heart failure and cardiogenic shock (35). Management involves hemodynamic support followed by coronary angioplasty or coronary bypass surgery. Unfortunately, the mortality rate in cardiogenic shock can be as high as 80% (36), but hospitals that provide timely PCI can expect 10% fewer deaths (36).
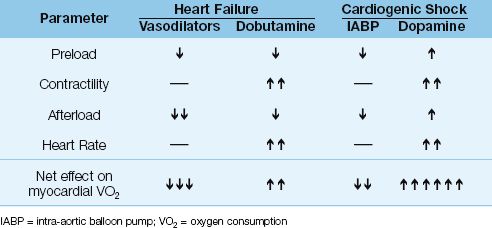
Hemodynamic Support
Hemodynamic support for acute heart failure and cardiogenic shock is described in Chapter 13. When these conditions are the result of coronary insufficiency, hemodynamic support should be designed to augment cardiac output without increasing cardiac work and myocardial oxygen consumption. Table 16.6 shows the effects of hemodynamic support measures on the determinants of myocardial O2 consumption (preload, contractility, afterload, and heart rate) for acute heart failure and cardiogenic shock. As judged by the net effect on myocardial O2 consumption, vasodilator therapy is the best choice for acute heart failure, and the intra-aortic balloon pump is the best choice for cardiogenic shock.
Reperfusion
Hemodynamic support is, of course, a bridge to interventions that re-establish flow in the infarct-related blood vessels. The ACC/AHA guidelines (3) recommend immediate PCI when cardiogenic shock appears within 36 hours of STEMI and when the angioplasty can be performed within 18 hours of the onset of shock. Coronary artery bypass surgery is considered if the cardiac catheterization reveals multivessel disease that is not amenable to angioplasty.
Table 16.6 Hemodynamic Support and Myocardial O2 Consumption
ACUTE AORTIC DISSECTION
Aortic dissection involving the ascending aorta can be mistaken as ACS, and can also be a cause of ACS. However, unlike ACS, aortic dissection is a surgical emergency that is often fatal if not managed appropriately.
Pathophysiology
Aortic dissection occurs when a tear in the aortic intima allows blood to dissect between the intimal and medial layers of the aorta, creating a false lumen. This process can be the result of atherosclerotic damage in the aorta from hypertension, or accelerated degradation of the aortic wall from a genetic disorder (e.g., Marfan’s syndrome). The dissection can originate in the ascending or descending aorta, and can propagate in antegrade and retrograde directions. When a dissection involves the ascending aorta, retrograde propagation can result in coronary insufficiency, aortic insufficiency, and pericardial tamponade, while antegrade propagation can result in neurologic deficits (from involvement of the aortic arch vessels) (37).
Clinical Manifestations
The most common complaint is the abrupt onset of chest pain. The pain is often sharp, and can be substernal (ascending aortic dissection) or in the back (descending aortic dissection). Most importantly, the chest pain can subside spontaneously for hours to days (39.40), and this can be a source of missed diagnoses. The return of the pain after a pain-free interval is often a sign of impending aortic rupture. About 5% of patients with acute aortic dissection are devoid of pain (37).
Clinical Findings
The most common clinical findings are hypertension (50% of patients) and aortic insufficiency (50% of patients) (38,39). Unequal pulses in the upper extremities (from obstruction of the left subclavian artery in the aortic arch) are found in as few as 15% of patients (39). The chest x-ray can show mediastinal widening (60% of cases) (39), but normal chest x-rays are reported in up to 20% of cases (37). The ECG can show ischemic changes (15% of cases) or evidence of MI (5% of cases), but the ECG is normal in 30% of cases (37). Because of the limited sensitivity of clinical findings, additional imaging studies are required for the diagnosis.
Diagnostic Tests
The diagnosis of aortic dissection requires one of four imaging modalities (40): magnetic resonance imaging (MRI) (sensitivity and specificity 98%), transesophageal echocardiography (sensitivity 98%, specificity 77%), contrast-enhanced computed tomography (sensitivity 94%, specificity 87%), and aortography (sensitivity 88%, specificity 94%). As indicated MRI is the most sensitive and specific imaging modality for the diagnosis aortic dissection. However, the immediate availability of MRI is limited in some hospitals, and CT angio-graphy is often the diagnostic test when aortic dissection is suspected.
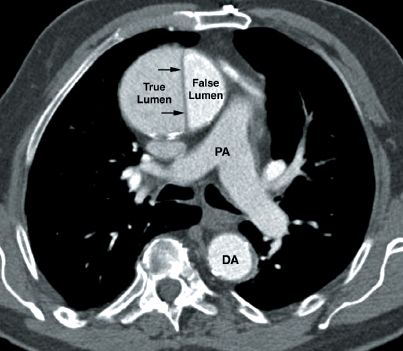
FIGURE 16.5 Contrast-enhanced CT image showing an aortic dissection involving the ascending aorta. The characteristic finding is the intimal flap that separates the true and false lumens (indicated by the small arrows). PA = main pulmonary artery, DA = descending aorta.
The CT image in Figure 16.5 shows an aortic dissection involving the ascending aorta. The small arrows point to the intimal flap that separates the dissecting blood in the wall of the aorta (false lumen) from blood in the aortic lumen (true lumen). The presence of this flap distinguishes an aortic dissection from a saccular aneurysm of the aorta.
Management
There are two essential management goals in aortic dissection: i.e., control of hypertension (to prevent aortic rupture) and prompt surgical correction.
Antihypertensive Therapy
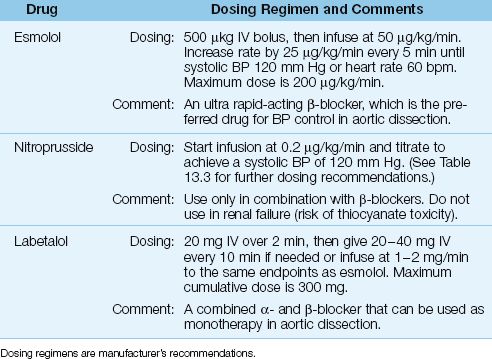
There is one major caveat for blood pressure control in aortic dissection: the reduction in blood pressure should not be accompanied by an increase in cardiac stroke output because increased flow in the aorta can promote further dissection. As a result, ?-receptor antagonists are preferred for blood pressure control in aortic dissection because of their ability to reduce the force of ventricular contraction (negative inotropic effect). The drug regimens used for blood pressure control in aortic dissection are presented in Table 16.7, and outlined below.
1. The β-blocker that is most favored is esmolol (Brevibloc), which has a short duration of action (9 minutes) and can be rapidly titrated to achieve the desired end-point (i.e., systolic blood pressure of 120 mm Hg and a heart rate of 60 bpm).
2. If the β-blocker does not achieve the desired effect, a vasodilator like nitroprusside can be added. Vasodilators should, however, never be used as monotherapy because they will increase cardiac output and increase the risk of progressive aortic dissection.
3. An alternative to dual therapy with a β-blocker and a vasodilator is the drug labetalol, which is a combined α-, β-Receptor antagonist that can be used as monotherapy for blood pressure control in aortic dissection.
With medical management alone, the mortality in acute aortic dissection increases 1–2% per hour after the onset of symptoms (37). Surgical repair reduces the mortality rate to 10% at 24 hours and 12% at 48 hours (37).
Table 16.7 Antihypertensive Therapy in Acute Aortic Dissection
A FINAL WORD
Blood Clots vs. O2 Transport
The discovery that acute myocardial infarctions are caused by blood clots that obstruct a coronary artery is at odds with the traditional teaching that global decreases in myocardial oxygen delivery (e.g., from anemia and hypoxemia) can promote ischemic injury in patients with coronary artery disease. This teaching is responsible for the current emphasis on blood transfusions and oxygen breathing to prevent myocardial ischemia in ICU patients with coronary artery disease. However, since blood clots cause heart attacks, not anemia or hypoxemia, these practices are unfounded.
Ever wonder why myocardial infarction is an uncommon occurrence during progressive circulatory shock and multiorgan failure (where myocardial O2 delivery is progressively threatened)? Now you have the answer.
REFERENCES
1. Roger V, Go AS, Lloyd-Jones D, et al. Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation 2012; 125:e2–e220.
Clinical Practice Guidelines
2. O’Connor RE, Brady W, Brooks SC, et al. Part 10: Acute coronary syndromes. 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation 2010; 122(Suppl 3):S787–S817.
3. Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction—executive summary. Circulation 2004; 110:588–636.
4. Kushner FG, Hand M, Smith SC Jr, et al. 2009 focused updates: ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction (updating the 2004 guideline and 2007 focused update) and ACC/AHA/SCAI guidelines for percutaneous coronary intervention (up-dating the 2005 guideline and the 2007 focused update). J Am Coll Cardiol 2009; 54:2205–2241.
5. Andersen JL, Adams CD, Antman EM. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction: executive summary. Circulation 2007; 116:803–877.
6. Jneid H, Anderson JL, Wright RS, et al. 2012 ACCF/AHA focused update of the guideline for the management of patients with unstable angina/non-ST-elevation myocardial infarction (updating the 2007 guideline and replacing the 2011 focused update). Circulation 2012; 212:1–36.
Reviews
7. Trost JC, Lange RA. Treatment of acute coronary syndrome: Part 1: non ST- segment acute coronary syndrome. Crit Care Med 2011; 39:2346–2353.
8. Trost JC, Lange RA. Treatment of acute coronary syndrome: Part 2: ST-segment elevation myocardial infarction. Crit Care Med 2012; 40:1939–1945.
Coronary Thrombosis
9. Davies MJ, Thomas AC. Plaque fissuring: the cause of acute myocardial in-farction. Br Heart J 1985; 53:363–373.
10. Van der Wal AC, Becker AE, van der Loos CM, Das PK. Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation 1994; 89:36–44.
11. Malek AM, Alper SL, Izumo S. Hemodynamic shear stress and its role in atherosclerosis. JAMA 1999; 282:2035–2042.
Routine Measures
12. Moradkan R, Sinoway LI. Revisiting the role of oxygen therapy in cardiac patients. J Am Coll Cardiol 2010; 56:1013–1016.
13. Bulkley GB. Reactive oxygen metabolites and reperfusion injury: aberrant triggering of reticuloendothelial function. Lancet 1994; 344:934–936.
14. Burls A, Cabello JB, Emperanza JI, et al. Oxygen therapy for acute myocardial infarction. A systematic review and meta-analysis. Emerg Med J 2011; 28:917–923.
15. Eikelboom JW, Hirsh J, Spencer FA, et al. Antiplatelet drugs. Chest 2012; 141 (Suppl): e89S–e119S.
16. ISIS-2 (Second International Study of Infarct Survival) collaborative group. Randomized trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 1988; 2:349–360.
17. Roux S, Christellar S, Ludin E. Effects of aspirin on coronary reocclusion and recurrent ischemia after thrombolysis: a meta-analysis. J Am Coll Cardiol 1992; 19:671–677.
Thrombolytic Therapy
18. Gruppo Italiano per lo Studio della Streptochinasi nell’Infarto Miocardico (GISSI). Effectiveness of intravenous thrombolytic treatment in acute myocardial infarction. Lancet 1986; 1:397–401.
19. Fibrinolytic Therapy Trialists Collaborative Group. Indications for fibrinolytic therapy in suspected acute myocardial infarction: collaborative overview of early mortality and major morbidity results from all randomized trials of more than 1000 patients. Lancet 1994; 343:311–322.
20. Boden WE, Kleiger RE, Gibson RS, et al. Electrocardiographic evolution of posterior acute myocardial infarction: importance of early precordial ST-segment depression. Am J Cardiol 1987; 59:782–787.
21. GUSTO Investigators. An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Engl J Med 1993; 329:673–682.
22. Smalling RW, Bode C, Kalbfleisch J, et al. More rapid, complete, and stable coronary thrombolysis with bolus administration of reteplase compared with alteplase infusion in acute myocardial infarction. Circulation 1995; 91:2725–2732.
23. GUSTO-III Investigators. An international, multicenter, randomized comparison of reteplase with alteplase for acute myocardial infarction. N Engl J Med 1997; 337:1118–1123.
24. Llevadot J, Giugliano RP, Antman EM. Bolus fibrinolytic therapy in acute myocardial infarction. JAMA 2001; 286:442–449.
25. Assessment of the Safety and Efficacy of a New Thrombolytic (ASSENT-2) Investigators. Single-bolus tenecteplase compared with front-loaded alteplase in acute myocardial infarction. Lancet 1999; 354:716–722.
26. Young GP, Hoffman JR. Thrombolytic therapy. Emerg Med Clin 1995; 13:735–759.
Percutaneous Coronary Intervention
27. The GUSTO IIb Angioplasty Substudy Investigators. A clinical trial comparing primary coronary angioplasty with tissue plasminogen activator for acute myocardial infarction. New Engl J Med 1997; 336:1621–1628.
28. Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomized trials. Lancet 2003; 361:13–20.
29. Stone GW, Cox D, Garcia E, et al. Normal flow (TIMI-3) before mechanical reperfusion therapy is an independent determinant of survival in acute myo-cardial infarction. Circulation 2001; 104:636–641.
30. Cannon CP, Gibson CM, Lambrew CT, et al. Relationship of symptom onset to balloon time and door-to-balloon time with mortality in patients undergoing angioplasty for acute myocardial infarction. JAMA 2000; 283:2941–2947.
31. Andersen HR, Nielsen TT, Rasmussen K, et al. for the DANAMI-2 Investi-gators. A comparison of coronary angioplasty with fibrinolytic therapy in acute myocardial infarction. New Engl J Med 2003; 349:733–742.
Adjunctive Therapies
32. Patrono C, C, Coller B, Fitzgerald G, et al. Platelet-active drugs: the relationship among dose, effectiveness, and side effects. Chest 2004; 126:234S–264S.
33. COMMIT (Clopidogrel and Metoprolol in Myocardial Infarction Trial) collaborative group. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomized placebo-controlled trial. Lancet 2005; 366:1607–1621.
Complications
34. Thompson CR, Buller CE, Sleeper LA, et al. Cardiogenic shock due to acute severe mitral regurgitation complicating acute myocardial infarction: a re-port from the SHOCK trial registry. J Am Coll Cardiol 2000; 36:1104–1109.
35. Samuels LF, Darze ES. Management of acute cardiogenic shock. Cardiol Clin 2003; 21:43–49.
36. Babaev A, Frederick PD, Pasta D, et al. Trends in the management and outcomes of patients with acute myocardial infarction complicated by cardiogenic shock. JAMA 2005; 294:448–454.
Aortic Dissection
37. Tsai TT, Nienaber CA, Eagle KA. Acute aortic syndromes. Circulation 2005; 112:3802–3813.
38. Khan IA, Nair CK. Clinical, diagnostic, and management perspectives of aortic dissection. Chest 2002; 122:311–328.
39. Knaut AL, Cleveland JC. Aortic emergencies. Emerg Med Clin N Am 2003; 21:817–845.
40. Zegel HG, Chmielewski S, Freiman DB. The imaging evaluation of thoracic aortic dissection. Appl Radiol 1995; (June):15–25.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


