Genetic factors
Environmental factors
Factor V Leiden
Surgery/trauma
Prothrombin gene mutation
Immobility
High factor VIII levels
Age
Hyperhomocysteinemia
Congestive heart failure/ myocardial infarction
Antithrombin deficiency
Hormone replacement therapy/oral contraception
Protein C deficiency
Cancer
Protein S deficiency
Pregnancy
Dysfibrinogenemia
Antiphospholipid antibodies
Air travel
The levels of thromboembolism risk are summarized in Table 14.2. Many procedures for trauma and orthopedic surgery have a high risk for VTE during and after the stay in the hospital. Importantly, the outpatient VTE rate is increased threefold when compared with inpatient VTE [3]. Almost half of the outpatients with VTE had been hospitalized in the recent past with a length of stay less than 4 days. Furthermore, less than half of the recently hospitalized patients had received appropriate VTE prophylaxis during their hospital stay [3], indicating a need for proper VTE prophylaxis in the surgical patient after hospitalization. Knudson et al. [1] found that in orthopedic and trauma surgery, 90 % of patients with VTE had one of the nine risk factors commonly associated with VTE. Six risk factors for VTE were found to be independently significant in multivariate logistic regression (Table 14.3).
Table 14.2
Levels of thromboembolism risk and recommended thromboprophylaxis in hospital patients
Levels of risk (VTE risk without thromboprophylaxis, %)a | Suggested thromboprophylaxis options |
|---|---|
Low risk: (up to 10 %)a | |
Minor surgery in mobile patients | No specific thromboprophylaxis |
Medical patients who are fully mobile | Early and “aggressive” ambulation |
Moderate risk: (10–40 %)a | |
Most general, open gynecologic or urologic surgery patients | LMWH (at recommended doses), LDUH bid or tid, fondaparinux |
Medical patients, bed rest or sick | |
Moderate VTE risk plus high bleeding risk (10–40 %)a | Mechanical thromboprophylaxisb |
High risk: (40–80) | |
Hip or knee arthroplasty, hip fracture surgery | LMWH (at recommended doses), fondaparinux, oral vitamin K antagonist (INR 2–3) |
Major trauma, spinal cord injury | |
High VTE risk plus high bleeding risk (40–80 %)a | Mechanical thromboprophylaxisb |
Table 14.3
Risk factors associated with VTE in trauma patients
Risk factor (number with risk) | Odds ratio (95 % CI) |
|---|---|
Age ≥40 years (n = 178,851)a | 2.29 (2.07–2.55) |
Pelvic fracture (n = 2,707) | 2.93 (2.01–4.27) |
Lower extremity fracture (n = 63,508)a | 3.16 (2.85–3.51) |
Spinal cord injury with paralysis (n = 2,852) | 3.39 (2.41–4.77) |
Head injury (AIS ≥3) (n = 52,197)a | 2.59 (2.31–2.90) |
Ventilator days >3 (n = 13,037)a | 10.62 (9.32–12.11) |
Venous injury (n = 1,450)a | 7.93 (5.83–10.78) |
Shock on admission (BP <90 mmHg) (n = 18,510) | 1.95 (1.62–2.34) |
Major surgical procedure (n = 73,974)a | 4.32 (3.91–4.77) |
A recent report sponsored by The Agency for Healthcare Research and Quality summarized a systematic review of 79 patient safety interventions based on the strength of the evidence: of the top ten safety practices, appropriate VTE for at-risk patients reached the highest rank. The others were perioperative beta-blockade, maximum sterile barriers for central venous catheter insertion, perioperative antibiotics to reduce postsurgical infections, patients restating/recalling what they have been told during informed consent, continuous aspiration of subglottic secretions to prevent ventilator-associated pneumonia, pressure-relieving bedding to prevent pressure ulcers, real-time ultrasonography to insert central venous catheters, self-management of warfarin, and nutritional support for postoperative and critically ill patients [4].
14.3 Prevalence
The prevalence of VTE in the absence of prophylaxis is shown in Table 14.4. Importantly, the incidence of VTE correlates with the number of risk factors [5].
Table 14.4
Prevalence of venous thromboembolism in the absence of prophylaxis
Medical | 10–20 % |
General surgery | 15–40 % |
Major gynecologic surgery | 15–40 % |
Major urologic surgery | 15–40 % |
Total hip, knee or hip fracture | 40–60 % |
Neurosurgery | 15–40 % |
Major trauma | 40–80 % |
Acute spinal cord injury | 60–80 % |
Stroke | 20–50 % |
Critical care | 10–80 % |
In 1975, Kakkar et al. [6] published a prospective, randomized, multicenter study of the effect of low-dose unfractionated heparin on fatal postoperative PE. They investigated 4,121 patients older than 40 years undergoing a variety of elective major surgical procedures and found a significant reduction of patients with fatal PE treated with 5,000 IU heparin (subcutaneously 2 h preoperatively and 8 h thereafter for 7 days) compared with the control group without treatment (0.097 % and 0.77 %, respectively). However, bleeding complications also were increased; they were not significant for excessive blood loss but were significant for the occurrence of wound hematoma.
14.4 Long-Term Effects and Outcome After VTE
14.4.1 Post-Thrombotic Syndrome
Despite appropriate anticoagulant therapy, at least one of every two or three patients with DVT of the lower extremities will develop post-thrombotic sequelae [7]. Prandoni et al. [8] studied the long-term outcomes after a first DVT. In a prospective cohort follow-up study, they analyzed 528 consecutive patients with a first episode of venography-confirmed DVT. Criteria for the post-thrombotic syndrome (PTS) were recorded and scored. The criteria to assess PTS included presence of leg symptoms (pain, cramps, heaviness, pruritus, and paresthesia) and clinical signs (pretibial edema, induration of the skin, hyperpigmentation, new venous ectasia, redness and pain during calf compression, and ulceration of the skin). In this study, the cumulative incidence of recurrent VTE was 17 % after 2 years, 24 % after 5 years, and 30 % after 8 years [8]. Malignancy and impaired coagulation inhibition increased the risk of recurrent VTE. In contrast, surgery and recent trauma or fracture was associated with a diminished risk of recurrent VTE. The cumulative incidence of PTS was 25 % after 2 years, 30 % after 5 years, and 30 % after 8 years. Importantly, the PTS occurred in almost one third of patients and was strongly related to recurrent ipsilateral DVT.
The factors potentially related to the development of the PTS are older age, obesity, a history of previous ipsilateral DVT, iliac-femoral location of the current thrombosis, failure to promptly recover from the acute symptoms, and insufficient quality of oral anticoagulant therapy. On the basis of recent findings, the lack of vein recanalization within the first 6 months after DVT seems to be an important predictor of PTS, while the role of venous reflux is controversial. According to the results of recent clinical studies, the prompt administration of adequate elastic compression stockings in patients with symptomatic DVT has the potential to reduce the frequency of PTS, and when carefully supervised and instructed to wear proper elastic stockings, more than 50 % of patients have the potential to either remain stable or improve during long-term follow-up. Nevertheless, because of limitations in current therapies, the management of PTS is demanding and often frustrating [7].
In a systematic review, Wille-Jørgensen et al. [9] studied the epidemiological association between asymptomatic postoperative DVT and the development of PTS. They found seven original publications (1984–2001) that addressed this question, mainly in abdominal surgery and hip and knee arthroplasty. The overall relative risk of developing PTS was 1.58 (95 % confidence interval, 1.24–2.02) in patients suffering from asymptomatic DVT compared to patients without DVT (P < 0.0005). They concluded that there is a significant association between postoperative asymptomatic DVT diagnosed with sensitive screening methods and occurrence of late PTS.
14.4.2 Pulmonary Hypertension
Pengo et al. [10] conducted a prospective, long-term follow-up study to assess the incidence of symptomatic chronic thromboembolic pulmonary hypertension (CTPH) in consecutive patients with an acute episode of PE but without prior venous thromboembolism. CTPH was considered to be present if systolic and mean pulmonary artery pressures exceeded 40 and 25 mmHg, respectively; pulmonary-capillary wedge pressure was normal; and there was angiographic evidence of disease. They found that symptomatic CTPH affects approximately 4 % of patients within 2 years after a first episode of symptomatic PE, with no subsequent increase in incidence. The following factors increased the risk of CTPH: a previous PE, younger age, a larger perfusion defect, and idiopathic PE at presentation.
14.4.3 Economic Burden
Although data on the exact cost attributed to VTE are lacking, a recent analysis of health care claims estimated that the total annual health care cost for VTE ranges from $7,594 to $16,644 per patient [11]. With estimates of 300,000–600,000 incident cases per year, this cost equates to a total annual cost of $2 to $10 billion attributable to VTE.
Dimick et al. [12] studied hospital costs associated with postoperative complications. The observational cohort study merged the National Surgical Quality Improvement Project private sector database to the hospital internal accounting data available in the University of Michigan Data Warehouse. The median total hospital costs (and the mean length of hospital stay) were $33,589 (20 days) for a patient with thromboembolic complications compared with $5,233 (5 days) for a patient without thromboembolic complication; $13,083 (9 days) for patients with infectious complications; $18,496 (4 days) for patients with cardiovascular complications; and $62,704 (19 days) for patients with respiratory complications.
An economic evaluation of VTE prophylaxis strategies in critically ill trauma patients at risk of bleeding has been published by Chiasson et al. [13]. They conclude that the attributable mortality resulting from PE in trauma patients with severe injuries is low relative to other causes of mortality. Prophylactic placement of a vena cava filter (VCF) in patients at high risk of VTE who cannot receive pharmacological prophylaxis is expensive and associated with an increased risk of DVT. Compared with the other strategies, serial Doppler ultrasound screening was associated with better clinical outcomes and lower costs [13].
14.5 Physiology of Plug Formation
Plug formation is a complex process that includes platelet aggregation and fibrin generation with the aim to generate a hemostatic clot. Platelet-plug formation at sites of injury occurs rapidly, within seconds of injury, and is of great importance to stop blood loss. The plasma coagulation system takes several minutes before this process is completed. The fibrin strands reinforce the platelet-induced plug. This reaction is particularly important in larger vessels to prevent recurrent bleeding hours or days after the initial injury. These two systems are closely linked. For example, activated platelets accelerate plasma coagulation and products of the plasma coagulation reaction, such as thrombin, stimulate platelet aggregation [14].
14.5.1 Coagulation
The coagulation pathways are a series of reactions in which inactive enzyme precursors and their co-factors are activated to become active components that then catalyze the next reaction in the cascade that ultimately results in cross-linked fibrin. Two pathways are known: (1) the contact activation (intrinsic) pathway, and (2) the tissue factor (extrinsic) pathway (Fig. 14.1).
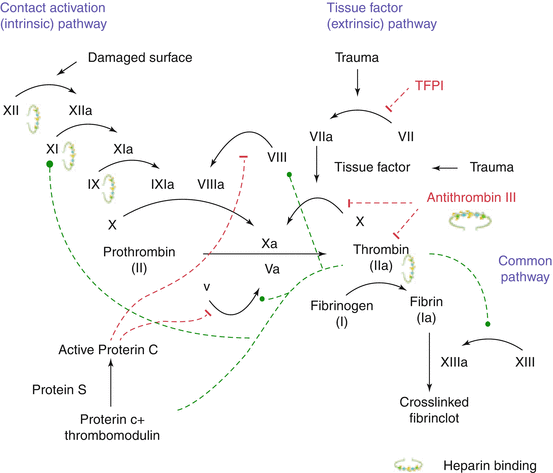
Fig. 14.1
Simplified model of the coagulation cascade. The extrinsic and intrinsic pathway merge in the activation of factor X, with further promotes the generation of thrombin and fibrin trough the common pathway. Heparin has unspecific binding to thrombin, IXa, XIa, and XIIa. The specific binding of heparin to antithrombin III eventually leads to binding of ATII to Xa with consecutive inactivation of Xa. Low molecular weight heparins and fonadaparinux also specifically bind to ATIII and thereby inactivate Xa. Activating effects are shown in dotted green lines, inhibiting effects are shown in dotted red lines. Abbreviations: Factors are abbreviated according to their number; a activated, ATIII antithrombin III, TFPI tissue factor pathway inhibiting factor (Adapted from [37])
The contact activation pathway begins at a damaged surface with formation of the primary complex on collagen by high-molecular-weight kininogen (HMWK), prekallikrein, and FXII (Hageman factor). Prekallikrein is converted to kallikrein and FXII becomes activated (FXIIa). The steps are demonstrated in Fig. 14.1. The minor role that the contact activation pathway has in initiating clot formation can be illustrated by the fact that patients with severe deficiencies of FXII, HMWK, and prekallikrein do not have a bleeding disorder.
The main role of the tissue factor pathway is to generate a “thrombin burst,” a process by which thrombin, the most important constituent of the coagulation cascade, is released instantaneously. FVIIa circulates in higher amounts than any other activated coagulation factor.
The final common pathway starts with the generation of thrombin. Thrombin has different functions. Its primary role is the conversion of fibrinogen to fibrin with consequent formation of a hemostatic plug. In addition, thrombin activates Factors VIII and V and their inhibitor protein C (in the presence of thrombomodulin), and thrombin activates Factor XIII, which forms covalent bonds that crosslink the fibrin polymers.
Following activation by the contact factor or tissue factor pathways, the coagulation cascade is maintained in a prothrombotic state by the continued activation of FVIII and FIX to form the tenase complex, until it is down-regulated by the anticoagulant pathways.
14.6 Monitoring of the Coagulation Cascade
14.6.1 The Prothrombin Time
Prothrombin time (PT) was discovered by Dr. A. Quick and colleagues in 1935, and a second method was published by Dr P. Owren, also called the “p and p” or “prothrombin and proconvertin” method. It was used as a measure of activity for warfarin when used therapeutically.
PT and its derived measures of prothrombin ratio and international normalized ratio (INR) are measures of the extrinsic pathway of coagulation (i.e., factors II, V, VII, X and fibrinogen).
PT is used to determine the clotting tendency of blood in order to estimate warfarin dosage, liver damage, and vitamin K status.
The reference range for prothrombin time is usually around 12–15 s.
The INR, that is now widely accepted worldwide, was introduced in the early 1980s when it turned out that there was a large degree of variation between the various PT assays. This discrepancy was mainly a result of problems with the purity of the thromboplastin (tissue factor) concentrate.
The normal range for the INR is 0.8–1.2.
14.6.2 Partial Thromboplastin Time
Partial thromboplastin time or activated partial thromboplastin time (aPTT) is a performance indicator that measures the efficacy of both the contact activation pathway and the common coagulation pathways. Apart from detecting abnormalities in blood clotting, it is also used to monitor the treatment effects with heparin.
14.6.3 Platelets
Platelets control bleeding when there is an injury to the blood vessel wall, and the endothelial cell layer is disrupted exposing the underlying extracellular matrix. Two adhesion receptors, glycoprotein (GP) Ib-IX–V that binds von Willebrand factor (vWF) and GPVI that binds collagen, are primarily responsible for regulating the initial platelet adhesion and activation in flowing blood. Following adhesion, rapid signal transduction leads to platelet activation, cytoskeletal changes associated with shape change, spreading and secretion, and inside–out activation of integrins that support adhesion and aggregation. The major platelet integrin GPIIb–IIIa binds vWF or fibrinogen and mediates platelet aggregation under shear conditions. Platelet activation that involves GPIb-IX–V or GPVI also leads to secretion of platelet agonists, such as adenosine diphosphate (ADP), which acts via the G protein-coupled receptors, P2Y1 and P2Y12, to reinforce platelet aggregation (Fig. 14.2). The activated platelet aggregates interact with the coagulation cascade and lead to stabilization of the clot by fibrin and GPIIb–IIIa-dependent mechanisms [15].
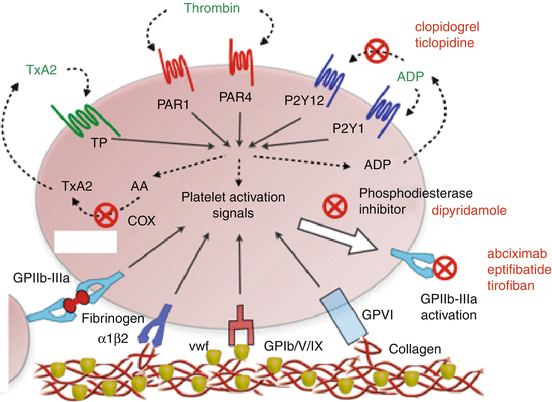
Fig. 14.2
Mechanisms of platelet activation and mode of action of current antiplatelet therapies. Platelets are initially activated by ligation of adhesion receptors by subendothelial proteins: glycoprotein (GP)VI and the integrin α2β1 bind collagens (red strands); GPIb/V/IX complex binds von Willebrand factor (vWf: olive spheres); and GPIIb–IIIa binds fibrinogen (red dumbbell). Propagation of platelet activation occurs by blood-soluble agonists: ADP released from activated platelets activates P2Y1 and P2Y12; and thromboxane A2 (TxA2) synthesised and released from activated platelets acts on thromboxane/prostanoid (TP) receptors; locally-generated thrombin activates protease-activated receptor (PAR)1 and PAR4. These inputs each signal the activation of GPIIb–IIIa, the key event in platelet aggregation. Antiplatelet agents inhibit the synthesis of TxA2 from arachidonic acid (AA) by the enzyme cyclooxygenase (COX)(aspirin), the binding of ADP to P2Y12 (clopidogrel and ticlopidine), the binding of ligands to activated GPIIb-IIIa (abciximab, eptifibatide and tirofiban), or the inhibition of intracellular signaling events mediated by phosphodiesterase (dipyridamole) (Adapted from [38, 39])
14.7 Mechanical Methods of Thromboprophylaxis
Early and frequent ambulation of hospitalized patients at risk for VTE is an important principle of patient care. However, many patients cannot be fully mobilized early after hospital admission or after surgery. In addition, mobilization alone does not provide adequate thromboprophylaxis for hospitalized patients and the majority of hospital-associated, symptomatic thromboembolic events occur after patients have started to ambulate.
Specific mechanical methods of thromboprophylaxis include graduated compression stockings (GCS), the venous foot pump (VFP), and intermittent pneumatic compression (IPC) devices. These methods increase venous outflow and/or reduce stasis within the leg veins and have important advantages and limitations (Table 14.5). The primary attraction of mechanical thromboprophylaxis is the lack of bleeding potential and the advantages for patients with high bleeding risks. While all three of the mechanical methods (GCS, VFP and IPC) reduce the risk of DVT in a number of patient groups, they have been studied less intensively than anticoagulant-based methods, and they are generally less efficacious when compared with anticoagulant thromboprophylaxis. Importantly, no mechanical thromboprophylaxis option has been studied in a large enough sample to determine if there is a reduction in the risk of death or PE.
Table 14.5
Advantages and limitations of mechanical thromboprophylaxis modalities
Advantages |
Can be used in patients at high bleeding risk |
Risk of bleeding is not increased |
Efficacy has been demonstrated in a number of patient groups |
May enhance the effectiveness of anticoagulant thromboprophylaxis |
May reduce leg swelling |
Limitations |
Not as intensively studied as pharmacologic thromboprophylaxis (fewer studies and smaller) |
Almost all mechanical thromboprophylaxis trials were unblended and therefore have a potential for bias in high-risk groups are less effective than anticoagulant thromboprophylaxis |
Many specific mechanical devices have never been assessed in any clinical trial |
No established standards for size, pressure, or physiologic features |
Greater effect in reducing calf DVT than proximal DVT |
Effect on PE and death unknown |
May reduce or delay the use of more effective anticoagulant thromboprophylaxis |
Compliance by patients and staff often poor |
Trials may overestimate the protection compared with routine use cost: associated with purchase, storage, dispensing, and cleaning of the devices, as well as ensuring optimal compliance |
14.8 Pharmacology: Antithrombotic Agents
The ideal antithrombotic agent should include the following properties: a high efficacy to safety index, a predictable dose response, a parenteral and oral application, a rapid onset of action, the availability of a safe antidote, no side effects, and minimal interactions. To characterize and rate this potentially ideal agent, it is equally important to choose appropriate and clinically relevant endpoints such as fatal or nonfatal PE, proximal DVT, and PTS. On the other hand, factors that have an impact on cost or patient safety and comfort, (e.g., relevant side effects, need for laboratory monitoring, or oral administration of the compound) must be considered.
14.8.1 Vitamin-K Dependent Clotting Factors, Coumarins
The coumarins or vitamin K antagonists (VKAs) have been the mainstay of oral anticoagulant therapy for more than 65 years. Their effectiveness has been established by well-designed clinical trials for the primary and secondary prevention of VTE. VKAs are challenging to use in clinical practice for the following reasons:
VKAs have a narrow therapeutic window.
VKAs exhibit considerable variability in dose response among patients because of genetic and other factors.
VKAs are subject to interactions with drugs and diet.
Their laboratory control is difficult to standardize.
Maintenance of a therapeutic level of anticoagulation requires a good understanding of the pharmacokinetics and pharmacodynamics of warfarin and good patient communication.
Coumarins inhibit the vitamin K-dependent synthesis of biologically active forms of the calcium-dependent clotting factors II, VII, IX and X (Figs. 14.1 and 14.3), as well as the regulatory factors protein C, protein S, and protein Z. Other proteins not involved in blood clotting, such as osteocalcin, or matrix Gla protein, may also be affected.
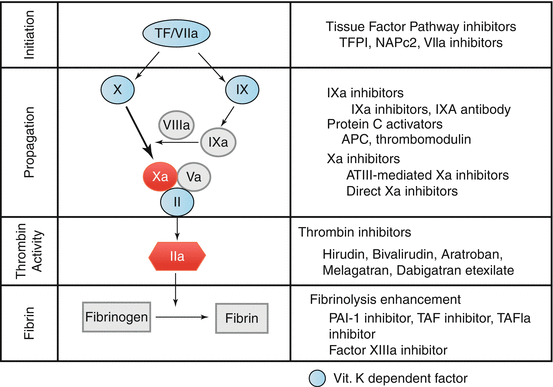
Fig. 14.3
Simplified model of the coagulation steps. Compared with coumarins that interfere with vitamin K dependent clotting factors new anticoagulant drugs have specific targets that interfere with the different steps of coagulation, modified according to [40, 41]. The important factors Xa and Thrombin are highlighted in red. The plasma half-life of the vitamin K dependent clotting factors (blue) are: VIIa 4–6 h, IX 24 h, X 48 h, and II 60 h. Abbreviations: TF tissue factor, II Prothrombin, IIa Thrombin, Va activated factor V, VIIa activated factor VII, IX factor IX, X factor X, APC activated protein C, TFPI tissue factor pathway inhibitor, NAPc2 Nematode Anticoagulant Protein c2
The antithrombotic effect of VKAs has conventionally been attributed to their anticoagulant effect, which in turn is mediated by the reduction of four vitamin K-dependent coagulation factors (Fig. 14.3). Evidence suggests, however, that the anticoagulant and antithrombotic effects can be dissociated and that the reduction of prothrombin and possibly factor X are more important than the reduction of factors VII and IX for the antithrombotic effect.
14.8.2 Heparin
Heparin is one of the oldest drugs that is currently still in widespread clinical use. Heparin’s discovery in 1916 can be attributed to the research activities of two men, Jay McLean and William Henry Howell. In the 1930s, Erik Jorpes discovered the structure of heparin. The first human trials of heparin began in 1935, and by 1937, heparin was considered to be a safe, easily available, and effective blood anticoagulant [16].
Heparin is a naturally occurring anticoagulant produced by basophils and mast cells. It was originally isolated from canine liver cells, hence its name (hepar is Greek for “liver”). Pharmaceutical grade heparin is derived from mucosal tissues of animals such as porcine intestine or bovine lung. Native heparin is a member of the glycosaminoglycan family of carbohydrates, which consists of variably sulfated repeating saccharide units. Interestingly, heparin has the highest negative charge density of any known biological molecule. Only one third of heparin molecules contain the high-affinity pentasaccharide required for anticoagulant activity.
Because of its negative charge, heparin binds in a nonspecific way to many proteins and other biological substances such as (1) receptors (steroids, growth factors receptors, ion channels); (2) enzymes (lipases, kinases, phosphatases); (3) cell matrix proteins (collagen, vitronectin, laminin); (4) nuclear proteins (histones, transcription factors); (5) lipoproteins (LDL, VLDL, apolipoproteins); (6) viral proteins (HIV, HSV, dengue); (7) prion protein, amyloid protein; (8) growth factors (FGF, VEGF); (9) heparin cofactor II; (10) thrombin, (11) enzymes Inhibitors (serpins), (12) Chemokines (PF4) (13) Heparin-binding protein HBP/CAP37 [17]. It is therefore not surprising that heparin, besides the desired anticoagulation effect, has many other biological effects that are related to size and nonspecific binding: thrombocytopenia (heparin-induced thrombocytopenia [HIT]), osteoporosis, endocrine and metabolic functions (glucose, lipid metabolism, calcium etc.), and modulation of inflammatory reactions [17, 18].
Specific binding of heparin to the enzyme inhibitor antithrombin III (ATIII) causes a conformational change that results in activation of ATIII. The activated ATIII then inactivates thrombin and other proteases involved in blood clotting, most notably factor Xa (Fig. 14.1). The rate of inactivation of these proteases by ATIII can increase by up to 1000-fold due to the binding of heparin. ATIII binds to a specific pentasaccharide sulfation sequence contained within the heparin polymer (Fig. 14.4): GlcNAc/NS(6S)-GlcA-GlcNS(3S,6S)-IdoA(2S)-GlcNS(6S). However, for thrombin inhibition, thrombin must also bind to the heparin polymer at a site proximal to the pentasaccharide. The highly-negative charge density of heparin contributes to its very strong electrostatic interaction with thrombin and represents a physiologically important nonspecific binding of heparin. The formation of a ternary complex between ATIII, thrombin, and heparin results in the inactivation of thrombin. For this reason heparin’s activity against thrombin is size-dependent, the ternary complex requires at least 18 saccharide units for efficient formation. In contrast, anti factor Xa activity only requires the specific pentasaccharide sequence for binding.
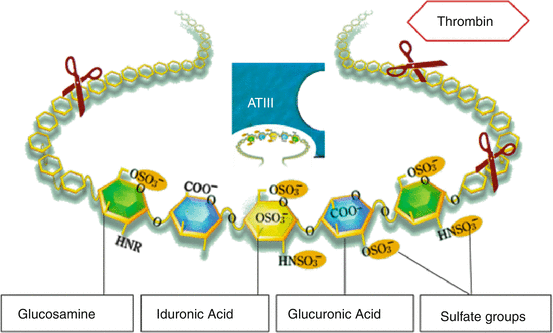
Fig. 14.4
Heparin. Heparin is a polysaccharide chain that consists of 18–70 monosaccharides. The scissors represent potential cutting sites during enzymatic or chemical processing of heparin to low molecular weight heparins (LMWH). The five monosaccharides GlcNAc/NS(6S)-GlcA-GlcNS(3S,6S)-IdoA(2S)-GlcNS(6S) provide the specific binding site to ATIII. New synthetic compounds such as fondaparinux consist of the specific ATIII binding sequence. For thrombin inhibition, however, thrombin must also bind to the heparin polymer at a site proximal to the pentasaccharide, this requires at least 18 saccharide units. This explains why LMWH have weaker and fondaparinux or no anti-thrombin activity. Abbreviations: ATIII antithrombin III (Adapted from [42])
14.8.3 Low-Molecular-Weight Heparin
In order to reduce the size-related and nonspecific effects of heparin, various methods of heparin processing have been developed [19]. Hence, low-molecular-weight heparin (LMWH) consists of only shorter chains of polysaccharide (Fig. 14.4). As might be expected, products prepared by distinctly different processes have different physical, chemical, and biological properties. The main differences between LMWH and “unfractioned” heparin are:
LMWH has a weaker effect on thrombin compared to heparin, but maintains the same effect on factor Xa.
Average molecular weight of LMWH is about 4.5 kDa, of heparin about 15 kDa.
Once-daily dosing by subcutaneous injection, rather than a continuous infusion of unfractionated heparin.
No need for monitoring of the coagulation parameter aPTT.
Possibly a smaller risk of bleeding.
Smaller risk of nonspecific effects such as risk of heparin-induced thrombocytopenia or osteoporosis in long-term use.
LMWH permits outpatient treatment of conditions such as DVT or PE that previously mandated inpatient hospitalization for unfractionated heparin administration, because LMWH can be given subcutaneously and does not require aPTT monitoring.
14.8.4 ATIII Mediated, Indirect Xa Inhibitors: Fondaparinux
In order to further reduce the size-related and nonspecific effects of heparin and LMWH, the group of Petitou, Herbert and colleagues [20] realized to synthesize the ATIII-binding sequence of heparin, the pentasaccharide fondaparinux. The main characteristics of fondaparinux are:
It catalyzes the inhibition of factor Xa, but not thrombin, in an antithrombin-dependent fashion.
It binds only to antithrombin; therefore, HIT and osteoporosis are unlikely to occur.
It has excellent bioavailability when administered subcutaneously, has a longer half-life than LMWHs.
It is administered once daily by subcutaneous injection in fixed doses, without anticoagulant monitoring.
In addition to the pharmacological anticoagulatory properties, the total chemical synthesis of fondaparinux also showed other advantages: (1) single chemical entity, (2) no risk of pathogen contamination, and (3) batch-to-batch consistency. Importantly, many clinical trials showed a significantly reduced VTE rate in major orthopedic surgery (total hip arthroplasty, total knee arthroplasty) and hip fracture surgery when compared with the LMWH enoxaparin [21].
14.8.5 Direct Xa Inhibitors
Direct factor Xa inhibitors include parenteral agents as well as several orally active drugs. All of the direct factor Xa inhibitors are small molecules that reversibly block the active site of factor Xa. The large number of new oral factor Xa inhibitors highlights the ongoing focus on development of oral anticoagulants that may replace VKAs such as warfarin.
14.8.6 Inhibition of Thrombin Activity
In contrast to indirect anticoagulants, which require a plasma cofactor to exert their activity, direct thrombin inhibitors have intrinsic activity because they bind to thrombin and block its enzymatic activity. The currently approved direct thrombin inhibitors are hirudin, bivalirudin, argatroban [22], and dabigatran etexilate.
Hirudin is a 65-amino acid polypeptide originally isolated from the salivary glands of the medicinal leech, Hirudo medicinalis. Hirudin is now available in recombinant forms. Two recombinant forms of hirudin, known as lepirudin and desirudin, are currently approved for clinical use. Lepirudin is licensed for treatment of thrombosis complicating HIT, whereas desirudin is approved in Europe for postoperative thromboprophylaxis in patients undergoing elective hip arthroplasty.
Bivalirudin is a 20-amino acid synthetic polypeptide, an analog of hirudin. Bivalirudin is licensed as an alternative to heparin in patients with HIT (with or without thrombosis) who require percutaneous coronary interventions. In contrast to hirudin, bivalirudin is not immunogenic. However, antibodies against hirudin can cross react with bivalirudin in vitro. The clinical consequences of this cross-reactivity are uncertain.
Argatroban is a competitive inhibitor of thrombin and binds noncovalently to the active site of thrombin to form a reversible complex. The plasma half-life of argatroban is 45 min. It is metabolized in the liver via the cytochrome P450 3A4/5 enzyme system. Consequently, argatroban must be used with caution in patients with hepatic dysfunction. However, because it is not renally excreted, argatroban is particularly useful in patients with HIT with severe renal impairment.
Ximelagatran is an oral anticoagulant that acts by direct and reversible inhibition of thrombin and has (or had) the potential to replace warfarin [23]. In the US, the presentation of ximelagatran clinical program to the Food and Drug Administration Cardiovascular and Renal drugs Advisory Committee (CRAC) in September 2004 and the non-approval of the new drug application highlighted surprisingly divergent analyses of the benefits and risks associated with this drug [24]. Consequently, Ximelagatran was taken from the market. Ximelagatran has predictable pharmacokinetics and pharmacodynamics and was used with a fixed dose without coagulation monitoring. It is rapidly converted to its active form melagatran that directly inhibits thrombin. A regimen with melagatran intravenously (IV) followed by oral ximelagatran was registered and launched in Europe in 2004 for the short-term prevention of VTE in patients undergoing knee or hip replacement surgery. The database consisted of a total of 30,698 subjects and included five phase III pivotal studies. During the advisory panel debate, widely divergent analyses of the benefits and risks of ximelagatran were presented. Ximelagatran hepatic toxicity was a key feature leading the CRAC to conclude that the benefit risk ratio of ximelagtran was unfavorable for the proposed indications.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


