CHAPTER 6 Regulation of Cardiac Output
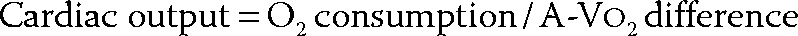
Arteriovenous Oxygen Difference
The provision of adequate tissue oxygenation depends on the integrated function of the heart, peripheral and pulmonary vasculature, lungs, blood, and peripheral metabolism.1 According to the Fick principle, the oxygen consumed by the body is equal to the product of the cardiac output and the A-VO2 difference. Under normal circumstances at rest, oxygen delivery exceeds consumption, so that adequate tissue oxygenation is provided with an A-VO2 difference of 40 ± 10 mL/L. If arterial oxygen tension and serum hemoglobin are normal, this results in a mixed venous oxygen saturation of 70% or more. If cardiac output decreases, the tissues extract a greater fraction of oxygen from the arterial blood, and mixed venous oxygen saturation decreases. A mixed venous oxygen saturation of 70% or more indicates that oxygen delivery and cardiac output are sufficient to meet the needs of the body.2
A wide A-VO2 difference and reduced mixed venous oxygen saturation may result from an abnormality of cardiovascular function that has resulted in a reduced cardiac output, a defect in blood oxygen-carrying capacity, or pulmonary disease. When the ability of the tissue to increase its extraction of oxygen is exhausted, tissue hypoxia results. In these conditions, anaerobic metabolism is heralded by a precipitous increase in venous lactate levels.3
Reflex Control of Cardiac Output
Under normal conditions, the heart has a large functional reserve; it is not the limiting factor in determining cardiac output. The arterial (perfusion) pressure and the cardiac output are adjusted to meet the needs of the body as they vary with posture and activity. The regulatory mechanisms involve sensory and effector components. The sensory components include peripheral receptors that react to changes in blood pressure (e.g., baroreceptors in aortic arch and carotid sinuses), blood volume (e.g., stretch receptors in the atria, Bainbridge reflex), and ventilation (e.g., carotid chemoreceptors). In addition there are loci in the cortex, hypothalamus, and diencephalon of the brain that react to emotions, anxiety, anticipation, exercise, hypoxia, and temperature. Cardiac output is modulated through changes in heart rate, stroke volume, and vasomotor tone that are mediated by direct parasympathetic and sympathetic neural pathways and by circulating catecholamines. Other humoral factors, such as adrenocortical steroids, thyroid hormones, insulin, and glucagons, have been shown to have an effect on cardiac function, but the importance of these hormones for regulation of cardiac output is unclear.
Direct sympathetic neural stimulation and circulating catecholamines exert a powerful stimulatory effect, increasing heart rate and contractile state, whereas vagal stimulation results in a decrease in heart rate and contractile state. The sympathetic and parasympathetic systems interact with each other in a complex fashion to influence cardiovascular performance. Generally, two types of interactions exist: accentuated antagonism and reciprocal excitation.4 Accentuated antagonism refers to the finding that the negative inotropic and chronotropic effects of vagal stimulation are more pronounced when vagal stimulation occurs in the presence of an increased adrenergic tone. Reciprocal excitation refers to the paradoxical effects of stimulation by one division on the autonomic nervous system, which results in effects normally expected from stimulation by the opposite autonomic division. The most common example of this is the production of positive inotropic effects by vagal stimulation or acetylcholine administration under experimental conditions.4
The factors that help regulate cardiac output are summarized in Table 6-1. The regulation system of cardiac output can become dysfunctional and result in syncope as a result of enhanced atrial and peripheral baroreceptor sensitivity, autonomic dysfunction, or complete heart block. In a critically ill cardiac patient, the normal regulatory mechanisms are usually saturated by maximal sympathetic and catecholamine stimulation. Under these conditions, the major determinants of cardiac output are no longer the neurohormonal pathways that regulate the normal cardiovascular system, but rather the interaction between the heart and the peripheral vasculature. The mechanical determinants of ventricular pump function are of paramount importance.
Table 6–1 Factors That Influence Cardiac Output
| Effects | |
|---|---|
| Sympathetic tone | ↑ contractile state, ↑ heart rate |
| Vagal tone | ↓ contractile state |
| Right vagus | ↓ sinus node activity, sinus bradycardia |
| Left vagus | ↓ atrioventricular conduction |
| Volume load | ↑ heart rate (Bainbridge reflex) |
| Baroreceptor stimulation (aortic arch, carotid sinus) | ↓ contractile state |
| Calcium administration | ↑ contractile state |
| Hormones (epinephrine, glucagon, thyroxine) | ↑ contractile state, ↑ heart rate |
| Drugs | |
| Positive inotropes | |
| Phosphodiesterase inhibitors (milrinone, amrinone, theophylline) | ↑ contractile state, ↑ heart rate |
| Digitalis glycosides | ↑ contractile state, ↓ atrioventricular conduction |
| Adrenergic stimulants (dopamine, dobutamine) | ↑ contractile state, ↑ heart rate |
| Negative inotropes | |
| β-adrenergic antagonists | ↓ contractile state, ↓ heart rate |
| Calcium channel blockers | ↓ contractile state, ↓ atrioventricular conduction |
Left Ventricular Performance
Pressure-Volume Loop
Although the integrity of left ventricular and right ventricular function and pulmonary and peripheral circulations is important, most cardiovascular dysfunction in adults is the result of impaired left ventricular function. The performance of the left ventricle can be understood by examining the relationship between left ventricular pressure and volume during a single beat in the pressure-volume plane (Fig. 6-1). Instantaneous intraventricular pressure is plotted on the y axis, and instantaneous ventricular volume is plotted on the x axis. At end-diastole (point a), ventricular pressure is relatively low, and ventricular volume is relatively high. The segment ab represents isovolumic contraction, with an increase in intraventricular pressure, but no ejection. Point b represents the start of ejection, coincident with the opening of the aortic valve when ventricular pressure exceeds aortic pressure. At end-systole (point c), the aortic valve closes, and a period of isovolumic relaxation commences (segment cd). The mitral valve opens at point d, when ventricular pressure decreases to less than atrial pressure, and ventricular filling commences.
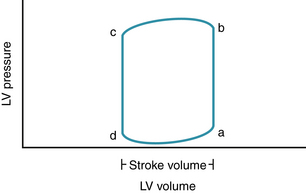
The difference between the end-diastolic and end-systolic volumes represents the volume ejected in that beat (stroke volume), and the ratio of stroke volume to end-diastolic volume is the ejection fraction. In the absence of aortic stenosis, the ventricular pressure at end-systole is the same as the pressure in the proximal aorta, and approximates systolic blood pressure (actually the pressure at the dicrotic notch in the aortic pressure-time course). Cardiac output is the product of stroke volume and heart rate. The pressure-volume loop provides a useful way to analyze the effects of contractile state, preload, and afterload on cardiac output.
Effect of Alterations in Preload on the Pressure-Volume Loop
Preload is defined as the stretch of the myocardium before activation and is readily indexed by end-diastolic volume. Within physiologic ranges, the greater the stretch on the myocardium, the stronger the ensuing contraction; this is known as the Frank-Starling relationship.5 From studies in isolated heart preparations in which preload, afterload, and contractile state were controlled, it has been shown that an increase in preload, produced by an increase in end-diastolic volume, results in an increase in the end-systolic pressure and the stroke volume of the ensuing beat.6–8
Three pressure-volume loops under three different preload conditions are shown in Figure 6-2. For the purpose of illustration, it is assumed that heart rate, contractile state, and afterload remain constant. Baseline conditions are represented by the shaded loop. A decrease in preload as a result of loss of blood volume, if not associated with any other change in afterload or contractile state, results in a smaller end-diastolic volume and a smaller pressure-volume loop that is shifted to the left. Conversely, a volume load results in a larger pressure-volume loop that is shifted to the right. An isolated increase in preload without any change in afterload or contractile state results in increases in stroke volume and end-systolic pressure if heart rate, afterload, and contractile state are unchanged. These conditions do not apply precisely in vivo. Isolated changes in preload, afterload, contractile state, or heart rate occur rarely because these changes are usually a response to, or in themselves result in, compensatory neurohormonal reflexes, which influence all these variables in a complex fashion. It may be useful, however, for an understanding of cardiovascular dynamics to analyze these factors separately.
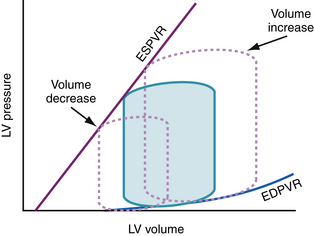
Figure 6-2
Related posts:
 Evolution of the Coronary Care Unit: Past, Present, and Future
Evolution of the Coronary Care Unit: Past, Present, and Future
 Elevated Cardiac Troponin in the Absence of Acute Coronary Syndromes: Mechanism, Significance, and Prognosis
Elevated Cardiac Troponin in the Absence of Acute Coronary Syndromes: Mechanism, Significance, and Prognosis
 Conduction Disturbances in Acute Myocardial Infarction
Conduction Disturbances in Acute Myocardial Infarction
 Pharmacologic Interactions in the CICU
Pharmacologic Interactions in the CICU
 Acute Aortic Syndromes: Diagnosis and Management
Acute Aortic Syndromes: Diagnosis and Management
 Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


