CHAPTER 33 Pulmonary Hypertension
Pulmonary Vascular Anatomy and Physiology
Starting from the main pulmonary arteries and proceeding distally towards the pulmonary capillaries, five structural regions can be identified.1 These include: (1) the large elastic arteries with a diameter greater than 3200 μm; (2) transitional arteries with diameters ranging from 2000 μm to 3200 μm; (3) muscular arteries with diameters ranging from 150 μm to 2000 μm; (4) partially muscularized arteries; and (5) nonmuscular arterioles with diameters ranging from 30 μm to 75 μm. The smallest muscular arteries and the partially muscularized arteries are felt to represent the resistance vessels of the pulmonary circulation.
The adult pulmonary circulation is a low resistance circulatory system that accommodates the whole cardiac output. Under normal circumstances, the resting pulmonary arterial pressure in man is fairly constant throughout life. The normal pulmonary artery pressure at sea level has a peak systolic value of 25 mm Hg, an end diastolic value of 10 mm Hg, and a mean value ranging from 12 to 16 mm Hg. By consensus, pulmonary hypertension is present when mean pulmonary arterial pressure exceeds 25 mm Hg at rest or 30 mm Hg with exercise.2 By definition, pulmonary hypertension is considered mild when the mean pulmonary artery pressure is 26 to 35 mm Hg, moderate when it is 36 to 45 mm Hg, and severe when it exceeds 45 mm Hg.
Exercise-related increases in pulmonary blood flow are associated with only minimal increases in pulmonary arterial pressure. As flow increases, therefore, pulmonary vascular resistance decreases. This response is primarily the result of recruitment of previously unperfused pulmonary vessels and, to a lesser extent, distention of already perfused vessels. Occasionally, especially in older subjects, pulmonary arterial pressure can increase to 50 mm Hg during exercise. This increase in pulmonary artery pressure is the result of an increase in pulmonary arterial wedge pressure, the probable result of decreased left ventricular diastolic compliance, rather than an increase in the pulmonary artery to pulmonary artery wedge pressure gradient.3
The hemodynamic evolution of pulmonary hypertensive disorders has been well described.4 Initially, the ability of the pulmonary vascular bed to dilate and to recruit unused vasculature is lost, resulting in increases in pulmonary artery pressure at rest and further elevations with exercise. In response to this increased afterload, the right ventricle undergoes hypertrophy. Early in the disease, the right ventricle is capable of sustaining a normal cardiac output at rest, although the ability to augment cardiac output with exercise is impaired. With disease progression, the right ventricle becomes incapable of maintaining a normal cardiac output at rest. As progressive right heart dysfunction develops, right heart diastolic and right atrial pressure increase, and right ventricular failure becomes clinically apparent. Although the left heart is not directly affected by pulmonary vascular disease, progressive right ventricular dilation can impair left ventricular compliance, leading to increased left ventricular end-diastolic and pulmonary capillary wedge pressures.
The main vascular changes in pulmonary arterial hypertension are vasoconstriction, smooth-muscle cell and endothelial-cell proliferation, and thrombosis. The endothelial cell plays a major role in the maintenance of normal pulmonary hemodynamics through the release of a variety of vasoactive substances, including nitric oxide (NO), prostanoids, and endothelin (ET), that contribute to vascular tone.5–7 Nitric oxide, previously known as endothelium-derived relaxing factor (EDRF), appears to play a central role in the modulation of pulmonary vascular tone. In addition to being a powerful vasodilator, nitric oxide acts as a bronchodilator, neurotransmitter, anticoagulant, antiproliferative, and antimicrobial substance and inhibits platelet aggregation. Continuous release of nitric oxide from pulmonary endothelium appears to determine the characteristically low pulmonary vascular resistance in man. Prostacyclin (PGI2), a product of cyclooxygenase metabolism of arachidonic acid, is a powerful vasodilator and inhibitor of platelet aggregation. Although it has been suggested that release of PGI2 by the endothelium modulates vasoconstriction, the continuous release of PGI2 does not appear necessary to maintain pulmonary arterial vasodilation. Endothelin, consisting of three discrete isoforms, represents one of the most powerful pulmonary arterial vasoconstrictors.
Classification of Pulmonary Hypertension
Pulmonary hypertension results when there is an absolute loss of pulmonary vascular cross-sectional area or an increased resistance to flow at any point within the pulmonary circulation. The pulmonary circulation is defined as extending from the pulmonic valve to the left atrium and consists of the pulmonary outflow tract, the right and left main pulmonary arteries and their lobar branches, the intrapulmonary arteries, the pulmonary arterioles, capillaries, venules, and large pulmonary veins. As a result, a diverse group of disease processes affecting any aspect of this circulation can result in pulmonary hypertension (Table 33-1). The term primary pulmonary hypertension (PPH) has been abandoned in favor of idiopathic pulmonary arterial hypertension (IPAH). The term pulmonary arterial hypertension (PAH) is reserved for those with a genetic basis for pulmonary hypertension, those with collagen vascular disease or other conditions associated with pulmonary arterial hypertension (APAH), and for the idiopathic variant (IPAH).
Table 33–1 Classification of Pulmonary Hypertension75
Pulmonary hypertension can result from a variety of obstructive and restrictive pulmonary parenchymal disorders.8–10 The basis for pulmonary hypertension in these disorders involves both an absolute loss of pulmonary cross-sectional area (bronchiectasis, emphysema) and, of greater importance, the chronic alveolar hypoxia, acidosis, and polycythemia that accompanies these disease states.
Independent of pulmonary parenchymal disorders, pulmonary hypertension is a frequent hemodynamic complication associated with a range of respiratory system disorders whose common physiologic abnormality are alveolar hypoxia and arterial hypoxemia of longstanding duration.11 Such disease processes include the obesity-hypoventilation syndrome, chest wall disorders, and neuromuscular disease.
There is also a group of disorders for which occlusion or narrowing of the large to medium-size pulmonary arteries is the site of increased resistance to flow. Included in this category is chronic thromboembolic disease, Takayasu arteritis, primary pulmonary artery tumors, and compression from mediastinal or hilar processes (carcinoma, fibrosis).12–15 Although it might be assumed that the progressive nature of the pulmonary hypertension in this group is primarily the result of the initiating disease process, there is increasing evidence that small vessel changes in the unobstructed vascular bed resulting from high pressure or flow, or mediator imbalance, may contribute significantly to the progression of the disease.16
Increased resistance to pulmonary venous drainage is a mechanism common to a number of diverse processes in which pulmonary hypertension occurs. Altered resistance to pulmonary venous drainage may be the result of diseases affecting the left atrium, the left ventricle (both systolic and diastolic), mitral or aortic valvular disease, or the large (mediastinal fibrosis) or small (pulmonary veno-occlusive disease) pulmonary veins. Additional vascular remodeling or vasoconstriction may contribute to the elevated pulmonary artery pressures and increased transpulmonary gradient, especially when left heart filling pressures have been markedly and protractedly increased.17
Finally, there is a group of disorders in which the association between pulmonary hypertension and the primary disease state remains obscure. Included among these is the pulmonary hypertension associated with HIV infection, and with cirrhosis and/or portal vein thrombosis.18–20
Clinical Presentation
The first challenge in the diagnostic evaluation of a patient with pulmonary hypertension is recognizing that pulmonary hypertension is, in fact, present. Diagnostic delay, especially in the absence of a history of an associated disease process, occurs commonly. Symptoms specifically attributed to the presence of pulmonary hypertension are nonspecific and dependent on the stage of the disease at which the patient presents (Table 33-2). A gradual decline in exercise tolerance and exertional dyspnea represent the initial manifestation and are present in essentially all patients with significant pulmonary hypertension. Other symptoms may include cough, chest pain, hemoptysis, and easy fatigability. These symptoms are also nonspecific and may lead the clinician, in the case of isolated pulmonary hypertension, to ascribe them to other causes (coronary artery disease, chronic obstructive pulmonary disease, psychogenic dyspnea); and, in the case of secondary pulmonary hypertension, to mistakenly ascribe them to a progression of the underlying disease.
Table 33–2 Functional Assessment of Patients with Pulmonary Hypertension76
| Class I: Patients with pulmonary hypertension but without resulting limitation of physical activity. Ordinary physical activity does not cause undue dyspnea or fatigue, chest pain, or near syncope. |
| Class II: Patients with pulmonary hypertension resulting in slight limitation of physical activity. These patients are comfortable at rest, but ordinary physical activity causes undue dyspnea or fatigue, chest pain, or near syncope. |
| Class III: Patients with pulmonary hypertension resulting in marked limitation of physical activity. These patients are comfortable at rest, but less than ordinary physical activity causes undue dyspnea or fatigue, chest pain, or near syncope. |
| Class IV: Patients with pulmonary hypertension resulting in inability to perform any physical activity without symptoms. These patients manifest signs of right heart failure. Dyspnea and/or fatigue may be present at rest, and discomfort is increased by any physical activity. |
One unique physical finding described in chronic thromboembolic pulmonary hypertension and congenital pulmonary artery stenosis is the presence of flow murmurs over the lung fields.21 The detection of these often subtle bruits, which appear to originate from turbulent flow through partially obstructed pulmonary arteries, can be instrumental in differentiating a large vessel from a small vessel etiology of pulmonary hypertension. These flow murmurs are detected over the lung fields rather than the precordium, are accentuated during inspiration, and are frequently heard only during periods of breath-holding.
Diagnostic Evaluation
In the evaluation of the patient with suspected pulmonary vascular disease, historical information should be gathered with the intent of identifying a disorder with a known association with pulmonary hypertension. In most instances, the presence or history of obstructive lung disease, restrictive lung disease, digital clubbing, obesity-hypoventilation syndrome, rheumatic fever, collagen vascular disease, HIV infection, or thromboembolism will antedate the occurrence of pulmonary hypertension and suggest a likely etiology. Intravenous drug use and the ingestion of certain anorexigenic medications (e.g., fenfluramine) have also been associated with the development of pulmonary hypertension.22–25 If the patient’s place of residence is a region endemic for histoplasmosis, this information is useful in suggesting the possibility of fibrosing mediastinitis. Although familial pulmonary hypertension is extremely rare, a family history of pulmonary hypertension should be explored with any individual having suspected pulmonary hypertension.26
Once an abnormality of the pulmonary vasculature has been considered as a basis for the patient’s complaints, the diagnostic sequence, despite the diverse diagnostic possibilities, is relatively straightforward if an orderly approach is followed (Fig. 33-1).
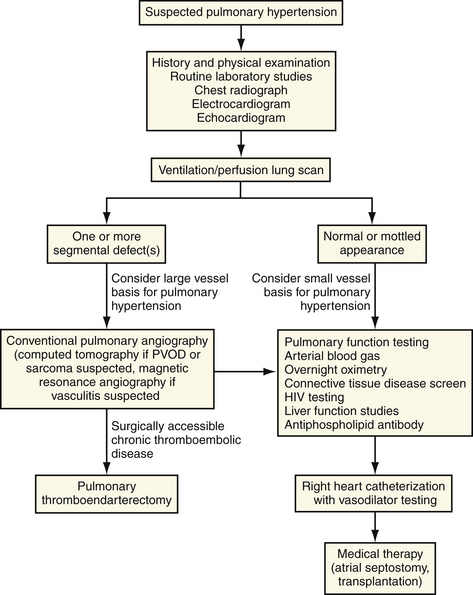
Findings on standard laboratory tests are dependent on the point in the natural history of the disease at which they are obtained. Abnormalities usually reflect the hemodynamic and gas exchange consequences of the pulmonary vascular obstruction and accompanying cardiac dysfunction. Electrocardiographic findings can suggest the possibility of pulmonary hypertension but are unlikely to differentiate among the diverse cardiac and pulmonary etiologies. Routine hematologic and blood chemistry studies are often unremarkable. Secondary polycythemia, resulting from long-standing hypoxemia, may be encountered. Abnormalities of liver function studies may be a consequence of hepatic congestion or may suggest the possibility of portopulmonary hypertension. Elevations in blood urea nitrogen (BUN) or uric acid levels may reflect the depressed cardiac output present in the latter stages of the disease.
Alterations in gas exchange may represent either an accompaniment of pulmonary hypertensive disorders or a causal factor. In disease processes not affecting the lung, chest wall, or respiratory control mechanism, hypoxemia is the consequence of a decreased mixed venous oxygen saturation resulting from a depressed cardiac output or from right-to-left shunting at either a cardiac or pulmonary level. In those processes which do affect the respiratory apparatus and which result in chronic hypoxia, vasoconstriction and vascular remodeling occur as a result of the hypoxemia, resulting in reduction of the pulmonary vascular bed to the degree necessary for the development of pulmonary hypertension.27 A central differential point is the presence of hypercarbia, which suggests advanced obstructive or restrictive parenchymal lung disease, disorders of the chest wall, or the presence of alveolar hypoventilation on either a central or neuromuscular basis.
Patients with chronic obstructive lung disease tend to have less severe pulmonary hypertension than other forms of pulmonary vascular disease although severe pulmonary hypertension may occur. The development of pulmonary hypertension in patients with chronic obstructive pulmonary disease represents a poor prognostic sign in terms of both mortality and quality of life. Mean pulmonary artery pressures rarely exceed 40 mm Hg, even in the presence of severe hypoxemia. This differs greatly from idiopathic pulmonary arterial hypertension, thromboembolic pulmonary hypertension. and from pulmonary hypertension associated with left heart and congenital heart diseases. In the latter stage of these disease processes, mean pulmonary artery pressure often exceeds 50 mm Hg, and can reach levels comparable to those of the systemic circulation. For this reason, pulmonary hypertension out of proportion to the degree of gas exchange impairment, or a rapid worsening of previously mild pulmonary hypertension in a patient with obstructive lung disease, should raise suspicion for a superimposed process such as thromboembolism, left ventricular dysfunction, or sleep apnea.28,29
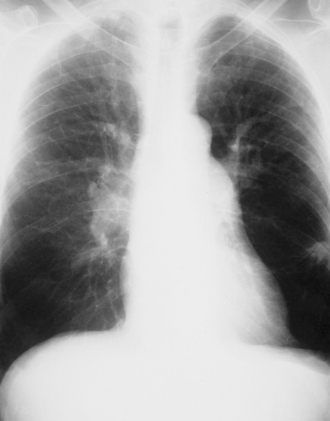
The chest radiograph may provide useful clues in the evaluation of a patient with suspected pulmonary hypertension (Fig. 33-2). If pulmonary hypertension is severe, enlargement of the right ventricle and pulmonary arteries (>16 mm enlargement of right interlobar pulmonary artery) may be recognized. Chest radiography can also be useful in suggesting the presence of pulmonary parenchymal, mediastinal, or cardiac problems responsible for the pulmonary hypertension.
Two noninvasive studies, echocardiography and ventilation-perfusion scanning, can provide valuable information regarding the severity and etiology of pulalmonary hypertension. Transthoracic echocardiography (TTE), although often technically difficult in patients with advanced lung disease, has evolved as an important noninvasive means of assessing the degree of pulmonary hypertension and right heart enlargement.30 Echocardiography has proved to be sensitive although somewhat less specific in detecting the presence of pulmonary hypertension in appropriate populations. Correlation between the echo-derived estimate of pulmonary artery pressure and that obtained at right heart catheterization is quite good within the range of pulmonary artery systolic pressures of 50 and 100 mm Hg.30,31
Related posts:
 Evolution of the Coronary Care Unit: Past, Present, and Future
Evolution of the Coronary Care Unit: Past, Present, and Future
 Elevated Cardiac Troponin in the Absence of Acute Coronary Syndromes: Mechanism, Significance, and Prognosis
Elevated Cardiac Troponin in the Absence of Acute Coronary Syndromes: Mechanism, Significance, and Prognosis
 Conduction Disturbances in Acute Myocardial Infarction
Conduction Disturbances in Acute Myocardial Infarction
 Antiplatelet Therapy
Antiplatelet Therapy
 Acute Aortic Syndromes: Diagnosis and Management
Acute Aortic Syndromes: Diagnosis and Management
 Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


