CHAPTER 42 Pharmacologic Interactions in the CICU
CONCOMITANT ADMINISTRATION of several drugs is common in the treatment of cardiovascular diseases. Often, combinations of drugs are necessary and result in increased efficacy of therapy, but with a large number of medications there is also an increased concern about drug interactions. Although the number of potential interactions is great, many are inconsequential. Conversely, drug interactions can have a significant adverse effect or even be lethal.1,2
Many drug interactions can be prevented by recognizing the drugs and the patients at risk. Situations with a high likelihood for clinically significant adverse drug interactions include the following1–4:
Mechanisms of interactions may be pharmacokinetic, affecting drug absorption, bioavailability, metabolism, or renal excretion; or pharmacodynamic, occurring at the sites of action in the heart such as sinoatrial and atrioventricular nodes, the intraventricular conduction system, and the smooth muscle.4
Vasodilators
Nitrates
The chief interactions of nitrates are pharmacodynamic. Nitrates (e.g., nitroglycerin, isosorbide dinitrate, isosorbide mononitrate) are widely used both in patients with angina and in those with congestive heart failure. A limitation in nitrate therapy is the development of tolerance, which is time-dependent.5 Many theories are proposed to explain this phenomenon6–8 including nitrate resistance,9 pseudotolerance (i.e., activation of counter-regulatory responses, such as secretion of catecholamines, angiotensin II, and endothelin)10 and “true” tolerance resulting from impaired bioconversion to nitric oxide (NO)11,12 and increased generation of superoxide.8
There are several reports of agents that limit or reverse nitrate tolerance when coadministered with nitroglycerin to isosorbide dinitrate. These include N-acetylcysteine, ACE inhibitors,13,14 angiotensin receptor blockers (ARBs),15 carvedilol,16 hydralazine,17 ascorbic acid,18 folic acid,19 and l-arginine.20 However, there are not enough data to make definite recommendations on drug combinations to prevent nitrate tolerance.
The combination of nitrates with hydralazine has been long known to be beneficial in congestive heart failure and is still used in patients who are intolerant of ACE inhibitors and in African-American patients who have reduced capacity for endogenous production of nitric oxide.21 These drugs may interact at the site of smooth muscle, involving inhibition of pyridoxal-dependent enzymes by hydralazine and resulting in an increased availability of sulfhydryl groups and prevention of nitrate tolerance by decreasing superoxide production and scavenging of reactive oxygen species.17,22
A clinically significant interaction occurs between nitrates and phosphodiesterase type 5 (PDE5) inhibitors, such as sildenafil and tadalafil, commonly used to treat erectile dysfunction.23–25 Inhibition of PDE5 results in increased cGMP levels, which are generated from endogenously derived NO. Nitrates exert their effect via biotransformation to NO and generation of cGMP. Coadministration of nitrates and PDE5 inhibitors causes significant reduction of blood pressure via a synergistic increase in cGMP levels, resulting in symptomatic hypotension and even death.23–25 PDE5 inhibitors are contraindicated for patients treated with nitrates. Conversely, nitrates should not be started within 24 hours of using sildenafil and 48 hours of using tadalafil.26
Another pharmacodynamic interaction occurs when nitrates are used with β-blockers, calcium channel blockers, or both as part of an intensive antianginal regimen, resulting in hypotension with reduced coronary flow. The result may be worsening of angina.27
Angiotensin-Converting Enzyme Inhibitors and Angiotensin Receptor Blockers
ACE inhibitors (Table 42-1) are widely used for the treatment of heart failure and hypertension, and to prevent remodeling after myocardial infarction.
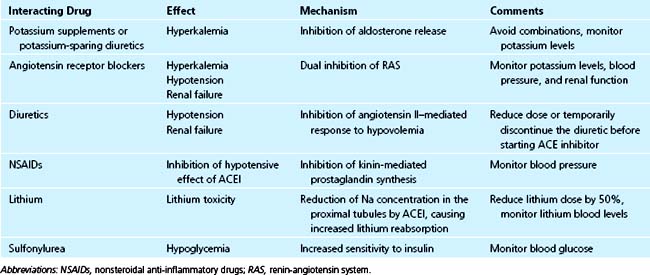
There is a beneficial effect in the synergism between ACE inhibitors and diuretics (e.g., thiazides and furosemide) in the treatment of hypertension and heart failure, but in a sodium-depleted patient or one with high renin levels, the combination can result in hypotension and worsening of renal failure. In such patients, the physician should start with a lower than normal dose of ACE inhibitor, temporarily lower the dose of diuretic, or discontinue the diuretic before administration of an ACE inhibitor.28 There is no evidence of a significant pharmacokinetic interaction between ACE inhibitors and diuretics.29 A synergistic effect on blood pressure reduction was also observed between various ACE inhibitors and calcium antagonists without pharmacokinetic interactions.28
No significant interactions were found between ACE inhibitors and β-blockers or digoxin.28,29
Among the interactions with noncardiovascular drugs, the most notable is with nonsteroidal anti-inflammatory drugs (NSAIDs) because of these drugs’ opposing effects on prostaglandin synthesis. The result is an attenuation of the antihypertensive effects of ACE inhibitors, predisposition to renal failure, or both.30,31 There are conflicting reports on the interactions between ACE inhibitors and low-dose aspirin.32,33 However, a retrospective analysis did not demonstrate an adverse effect of aspirin on the survival of patients with left ventricular systolic dysfunction treated with ACE inhibitors.34
There are reports of lithium toxicity when patients on chronic lithium therapy were started on ACE inhibitors. Because renal excretion of lithium is dependent on glomerular filtration and on sodium concentration in the proximal tubule, the possible mechanism of interaction may be the reduction of both by ACE inhibitors, especially in volume-depleted patients.28,29
A life-threatening anaphylactoid reaction has been described in a patient treated with ACE inhibitors while on hemodialysis with a polyacrylonitrile membrane (AN69). The possible mechanism of this interaction is activation of the kinin-kallikrein system by the surface of the AN69 membrane, resulting in an increased production of bradykinin, the breakdown of which is inhibited by ACE inhibitors.28 No adverse reactions were reported with other dialysis membranes.
ACE inhibitors increase insulin sensitivity, and there have been several reports of hypoglycemia when captopril or enalapril was given to patients receiving glibenclamide, although others did not observe this effect.35–37 It seems prudent to monitor for possible hypoglycemia when ACE inhibitors are given to patients who are already receiving oral antihypoglycemic agents.
Angiotensin receptor blockers (ARBs) have low potential for interaction with other drugs. Among ARBs, only losartan and irbesartan undergo significant metabolism by cytochrome P-450 enzymes (CYP 2C9 and CYP 3A4). Rifampin significantly reduces the AUC and the half-life of losartan and of its active metabolite, and dose increase of losartan may be indicated during concomitant administration.38 However, no clinically significant interactions were found between losartan and erythromycin, digoxin, or warfarin, and between irbesartan and digoxin, nifedipine, or hydrochlorothiazide.39 There was a report of a significant interaction between valsartan and lithium resulting in lithium toxicity.40
Attenuation of the hypertensive effect of ARBs may result from concomitant administration of NSAIDs, via mechanisms similar to the interaction with ACE inhibitors.39
Combination of ACE inhibitors and ARBs provides a dual and more effective blockade of the renin-angiotensin system (RAS). The combination was shown to be more effective in patients with diabetic and nondiabetic nephropathy and in patients with congestive heart failure.41,42 However, there are more adverse effects of hypotension, hyperkalemia, and renal dysfunction in patients on combination therapy.42 There are conflicting data on the use of dual RAS blockade with β-blockers in patients with CHF. Val-HeFT trial suggested an increased mortality in patients treated with triple therapy compared with an ACE inhibitor and β-blocker alone, while the CHARM and VALIANT trials did not find any increase in mortality in patients on triple therapy.43–45 The combination therapy of ACE inhibitor and ARB seems prudent in patients with CHF with highly activated RAS despite full-dose ACE inhibition, and in diabetic patients with nephropathy.41
Inotropic Drugs
Dopamine and Dobutamine
Vasoactive amines are used in the CICU to treat heart failure and shock. Both drugs are metabolized in the liver: dopamine by catechol-O-methyltransferase and monoamine oxidase; dobutamine by catechol-O-methyltransferase. Dopamine is inactivated in alkaline pH and therefore should not be administered in the same infusion as bicarbonate.46
Low-dose dopamine prevented norepinephrine-induced decreases in renal plasma flow in healthy volunteers.47 However, it is not clear whether the same effect is found in critical-care patients. A multicenter study of low-dose dopamine use in critical care patients did not find any improvement in renal outcomes, and there were more adverse effects in patients on dopamine.48,49
There is a report of interaction between dobutamine and low-dose carvedilol resulting in severe hypotension.50 The proposed mechanism is a fall in systemic vascular resistance due to excessive β2-receptor activation caused by a selective β1-receptor blockade by low-dose carvedilol. This interaction may be expected with other selective β1-blockers.
Digoxin
Digoxin is a drug with a narrow therapeutic range and is subject to many drug interactions, both pharmacokinetic and pharmacodynamic (Table 42-2).
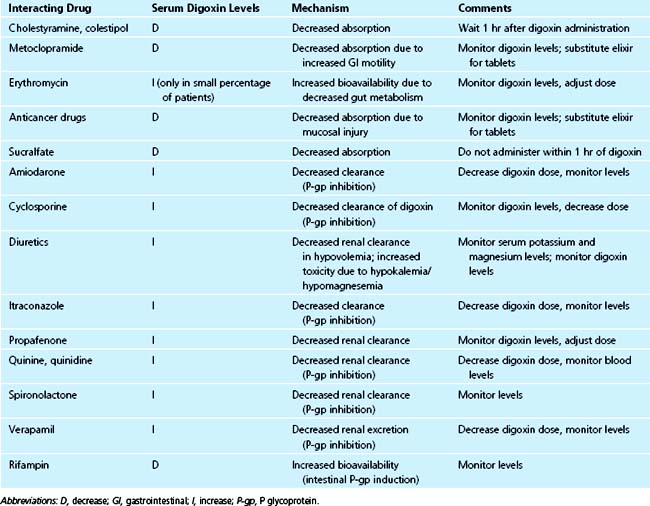
Interactions Affecting Absorption and Bioavailability
Digoxin tablets are absorbed slowly; therefore, agents that increase gastrointestinal motility (e.g., metoclopramide) may decrease its absorption, whereas agents that slow gastrointestinal transit (e.g., propantheline, other anticholinergic agents) may increase its absorption.51 Elixir preparations are usually not subject to these interactions because their absorption is more rapid.
Treatment with high-dose chemotherapeutic agents resulting in intestinal mucosal injury can reduce digoxin absorption from tablets by as much as 50% but does not significantly affect absorption from elixir.52
In about 10% of patients, digoxin that is not absorbed in the upper gastrointestinal tract or that is excreted in the bile is reductively metabolized by the anaerobic bacteria Eubacterium Lentum, which is part of the normal flora of the colon. Such metabolism can account for about 40% of digoxin elimination. These patients may be recognized by the characteristic of needing higher than usual doses of digoxin to achieve therapeutic levels. In this group of patients, treatment with broad-spectrum antibiotics (e.g., erythromycin, tetracycline) can result in significantly increased bioavailability and digoxin toxicity.53
Cholesterol-binding resins (e.g., cholestyramine, colestipol) bind digoxin in the gut and may reduce its absorption by 20% to 30%. This effect can be avoided by giving digoxin at least 1 hour before the resins.54
Sucralfate has been reported to decrease the absorption of digoxin.55 No significant interaction has been found between digoxin and antacids.56
Intestinal P-glycoprotein (P-gp) plays an important role in bioavailability of digoxin.57 Rifampin increases P-gp content in the intestine and decreases digoxin bioavailability, resulting in significantly lower digoxin AUC after oral administration.58 On the other hand, drugs that inhibit intestinal P-gp increase digoxin bioavailability and AUC. Dipyridamole was shown to increase digoxin bioavailability in vitro and in vivo; however, the effect on AUC was slight and clinically insignificant.59 Carvedilol significantly decreased oral clearance of digoxin in children, resulting in digoxin toxicity in some cases.60 However, in adults carvedilol caused only a modest increase in digoxin bioavailability and AUC.61
St. John’s wort (Hypericum perforatum) is an herbal medicine frequently used for treatment of depression. Hypericum induces intestinal P-gp, resulting in a 1.4-fold increased expression of duodenal P-gp in humans.62 Coadministration of St. John’s wort and digoxin caused a 25% decrease in AUC and a 33% reduction in trough and Cmax concentrations of digoxin.63
Interactions Affecting Elimination
Digoxin is eliminated primarily by renal excretion via the ATP-dependent efflux pump P-glycoprotein (P-gp).57,64–66 Basic drugs, among them amiodarone, clarithromycin, itraconazole, quinine, quinidine, verapamil, spironolactone, cyclosporine A, propafenone, and ritonavir decrease the renal clearance of digoxin by inhibiting P-gp in the kidney.67–72 Carvedilol caused significant increases in digoxin AUC and Cmax in men, but not in women.73 The explanation may be that men have a higher P-gp activity compared to women, and thus are more sensitive to the effects of inhibiting drugs.
Because the combination of digoxin with drugs that inhibit its elimination results in increased toxicity, the physician should reduce the digoxin dose by 50% when starting another drug such as quinidine or amiodarone. Because of the long half-life of digoxin, even after dose reduction there is still a potential for toxicity during the first week after adding another drug to the regimen, and patients must be monitored closely during this period.
Pharmacodynamic Interactions
Digitalis effect and toxicity are enhanced in the presence of hypokalemia and hypomagnesemia, and toxicity may be present even with digoxin blood levels within the therapeutic range. Because digoxin is often used with diuretics, which can cause electrolyte abnormalities, it is important to monitor and correct deficiencies, even though potassium and magnesium blood levels do not always accurately reflect body stores.74 Concomitant administration of digoxin and sympathomimetic or vagolytic drugs may mask digitalis toxicity because of opposing effects on cardiac conduction. This effect is used therapeutically when digoxin is administered at the beginning of quinidine therapy to prevent rapid atrioventricular node conduction as a result of the vagolytic effect of quinidine.
Antiarrhythmic Drugs
Drug interactions with antiarrhythmic drugs are presented in Table 42-3. A detailed review was published by Trujillo and Nolan.75
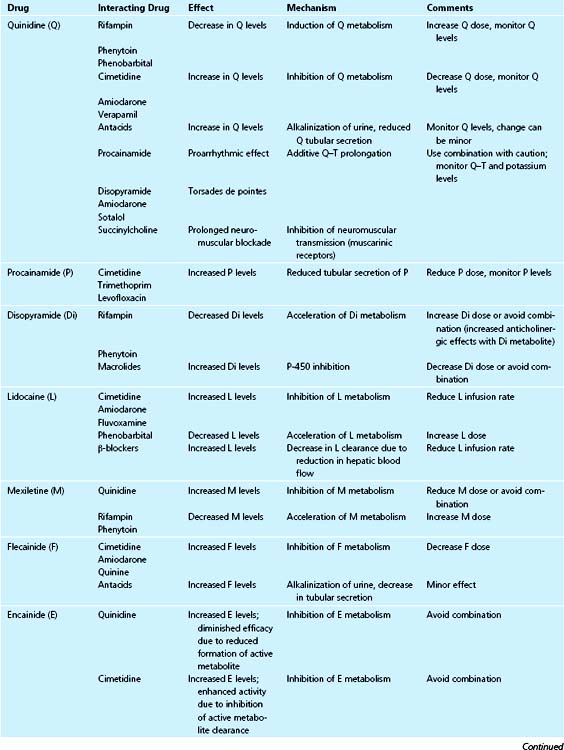
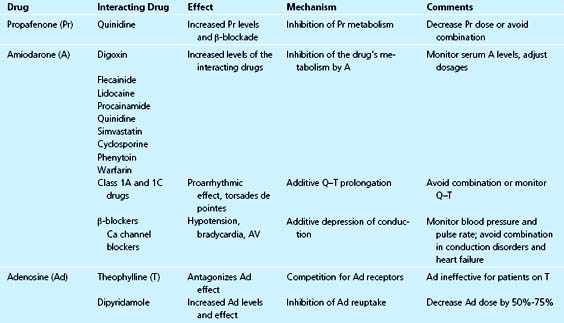
Class 1A
Quinidine
Quinidine is a drug with many adverse side effects, and it interacts with many other drugs.
Pharmacokinetic Interactions
Quinidine is primarily metabolized in the liver, and only about 20% is excreted in urine. Thus it is subject to interactions with drugs that either induce or inhibit liver enzymes. Among the enzyme inducers, phenobarbital, phenytoin, and rifampin accelerate the metabolism of quinidine, resulting in decreased blood levels. Therefore, when any of these drugs is added, the quinidine dose should be increased, and when these drugs are stopped, the quinidine dose should be decreased. The dose adjustment may be as great as threefold.76
There have been conflicting reports about the effect of nifedipine on quinidine blood levels.77 However, quinidine pharmacokinetics do not appear to be significantly changed by nifedipine or felodipine.77
Cimetidine, verapamil, and amiodarone inhibit quinidine metabolism and necessitate downward dose adjustment.78–80
Even though renal excretion accounts for only 20% of quinidine clearance, clearance is influenced by urine pH, and alkalinization of urine (e.g., with intensive antacid therapy) may result in a moderate increase in quinidine levels.81
Pharmacodynamic Interactions
The combination of quinidine with other class 1A drugs (e.g., disopyramide, procainamide) or with class 3 drugs (e.g., amiodarone, sotalol) can result in Q–T prolongation and increased risk of torsades de pointes.82,83
Moxifloxacin, a methoxyquinolone antibiotic, prolongs Q–T interval, and must be used with caution with class 1A or class 3 drugs.84 Hypokalemia, hypomagnesemia, or both, common with diuretic treatment, can increase Q–T prolongation and the risk of torsades de pointes from quinidine or other drugs.85
A proarrhythmic effect of combining amiloride and quinidine together has been described, probably resulting from a synergistic increase in sodium channel blockade.86
Quinidine has also been reported to potentiate the anticoagulant effects of warfarin by direct inhibition of clotting factor synthesis in the liver.81,87 Quinidine inhibits neuromuscular transmission and prolongs the duration of anesthesia when used with curare or succinylcholine.88
Procainamide
Procainamide is both metabolized and excreted renally, and its active metabolite, N-acetyl procainamide (NAPA), is cleared primarily by renal excretion. The major mechanism is tubular secretion with little reabsorption; therefore, urine pH changes do not cause significant changes in blood procainamide concentration. Conversely, other basic drugs that are secreted by renal tubules (e.g., cimetidine, ranitidine, trimethoprim) significantly inhibit procainamide secretion.89,90 Levofloxacin, but not ciprofloxacin, was found to significantly inhibit renal clearance of procainamide and of NAPA.91
Disopyramide
Disopyramide is metabolized by liver enzymes. Enzyme inducers (e.g., rifampin, phenytoin, barbiturates) enhance the metabolism of disopyramide and may cause subtherapeutic blood levels.92 Enzyme inhibitors such as cimetidine are expected to decrease disopyramide clearance and to increase blood levels. Macrolide antibiotics, including erythromycin, clarithromycin, and azithromycin, inhibit P450 enzymes and there are reports of disopyramide toxicity during concomitant administration.93–95
Class 1B
Lidocaine
Lidocaine is extensively metabolized by the liver, with a high extraction ratio; therefore, its clearance is dependent on hepatic blood flow. Lidocaine is usually administered as a bolus followed by continuous intravenous infusion. The distribution of a bolus to the tissues is slowed in patients with severe heart failure and shock and may result in high blood levels and toxicity.96 To avoid this, a bolus dose of lidocaine in these situations should be given slowly.
Drugs that decrease hepatic blood flow (e.g., β-blockers) decrease lidocaine clearance, whereas drugs that increase hepatic blood flow (e.g., dopamine and dobutamine) can be expected to increase clearance.97
Lidocaine clearance changes with liver enzyme inducers and inhibitors; it decreases with cimetidine and increases with phenobarbital.98 Amiodarone has been reported to reduce lidocaine clearance, resulting in high lidocaine plasma levels and seizures.99 Fluvoxamine, a CYP 1A2 inhibitor, significantly reduced lidocaine clearance.100 An interaction with mexiletine, resulting in increase in plasma lidocaine levels and toxicity, was reported.101 The purported mechanism is displacement of lidocaine from tissue binding sites. It is recommended that the rate of lidocaine infusion be adjusted by 25% to 50% in the presence of potential drug interactions.
Increased cardiotoxicity resulting in sinoatrial block has been reported with concomitant use of lidocaine and quinidine or procainamide.81,102
Mexiletine
Mexiletine is extensively metabolized by the liver, mainly by cytochrome P-450 isozyme 2D6, the activity of which is genetically determined, and in part by isozyme 1A2. Mexiletine metabolism is inhibited by quinidine.103 Fluvoxamine, a CYP 1A2 inhibitor, caused significant increases in mexiletine AUC.104 The enzyme inducers rifampin and phenytoin have been shown to enhance its metabolism.105
Class 1C
Flecainide
Flecainide is eliminated by renal excretion and hepatic metabolism. Changes in urine pH are expected to affect its clearance; alkalinization of urine will result in higher plasma concentrations. Cimetidine, quinine, and amiodarone have been reported to inhibit the clearance and increase plasma concentrations of flecainide.106,107
Encainide
Encainide is extensively metabolized by the liver with significant first-pass effect, and its metabolism is genetically determined. Its metabolites are active and are responsible for the majority of its pharmacologic activity.108 Quinidine inhibits encainide metabolism, resulting in diminished pharmacologic activity.109 Cimetidine inhibits the metabolism of the active metabolites of encainide even more than it inhibits encainide metabolism, resulting in enhanced pharmacologic activity.110
Propafenone
Propafenone is metabolized by cytochrome P-450 isozyme 2D6; thus its metabolism is subject to genetic polymorphism of 2D6; about 10% of whites are poor metabolizers of this drug. When propafenone is administered to extensive metabolizers, quinidine, a potent inhibitor of 2D6 enzyme, inhibits its clearance, resulting in higher blood levels and a significant increase in pharmacologic effect.111,112 Conversely, rifampin, a P-450 inducer, increases propafenone clearance.113 Propafenone has been reported to inhibit the metabolism of theophylline.114
Class 3
Amiodarone
Amiodarone has a number of significant pharmacokinetic and pharmacodynamic drug interactions.
Pharmacokinetic Interactions
Amiodarone is an inhibitor of certain cytochrome P-450 enzymes. Amiodarone potentiates the effect of warfarin and causes enhanced anticoagulation.115,116 Because the half-life of amiodarone is very long, this effect may take up to several weeks to reach maximum intensity. Conversely, after amiodarone is discontinued, its effect on warfarin metabolism can persist for several months. Amiodarone also elevates the blood levels of digoxin, quinidine, procainamide, flecainide, lidocaine, metoprolol, simvastatin, cyclosporine, and phenytoin.99,117–120 It is recommended that dosages of warfarin and digoxin should be decreased by half when amiodarone is added.75
Pharmacodynamic Interactions
The combination of amiodarone with class 1A or 1C drugs can result in enhanced antiarrhythmic efficacy. However, when combined with class 1A drugs such as quinidine, amiodarone can result in marked Q–T prolongation and an increased risk of torsades de pointes.74 Another pharmacodynamic interaction with calcium antagonists or β-blockers may result in sinoatrial node depression, with excessive bradycardia or sinus arrest.121 This effect may persist for weeks after discontinuing amiodarone. However, the combination of amiodarone and β-blockers is usually safe, and may be beneficial in post–MI patients.122
Sotalol
The administration of aluminum and magnesium hydroxide decreased sotalol absorption.123 Separation of the drugs by 2 hours avoided the interaction. Sotalol is excreted primarily unchanged by the kidneys. The same pharmacodynamic effects for the combination with class 1A drugs would be expected for sotalol as with amiodarone. Hypokalemia can aggravate the risk of torsades de pointes with this combination.124 Torsades de pointes was reported after the combination of sotalol and terfenadine.125 The combination of sotalol with other β-blockers may produce bradycardia, atrioventricular block, or exacerbation of heart failure.126
Class 4
Adenosine
Adenosine is used for acute treatment of supraventricular tachycardia. Dipyridamole inhibits adenosine reuptake, causing increased blood concentrations. Patients on dipyridamole should receive one fourth the standard dose of adenosine. Theophylline competes with adenosine for the same receptors and thus inhibits its action.127
Calcium Channel Blockers
The most commonly used calcium channel blockers (Table 42-4) (e.g., verapamil, diltiazem, nifedipine) are extensively metabolized by the liver; therefore, their metabolism is dependent on hepatic blood flow. Because of their extensive hepatic metabolism, they undergo significant first-pass metabolism in the liver, with a low level of bioavailability.128 Verapamil and diltiazem also inhibit some P-450 enzymes, thus affecting the metabolism of other drugs.
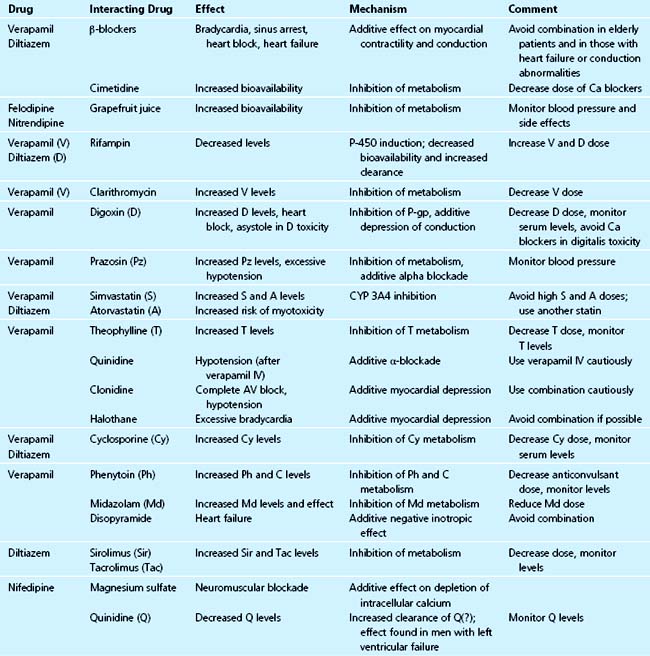
Pharmacokinetic Interactions
Cytochrome P-450 enzyme inducers (e.g., rifampin, phenytoin, phenobarbital) decrease the bioavailability and increase the clearance of verapamil and diltiazem. St. John’s wort also significantly decreases verapamil bioavailability through induction of first-pass metabolism in the gut.129 Conversely, the enzyme inhibitor cimetidine increases the bioavailability and decreases the clearance of calcium antagonists.130–132
Related posts:
 Evolution of the Coronary Care Unit: Past, Present, and Future
Evolution of the Coronary Care Unit: Past, Present, and Future
 Elevated Cardiac Troponin in the Absence of Acute Coronary Syndromes: Mechanism, Significance, and Prognosis
Elevated Cardiac Troponin in the Absence of Acute Coronary Syndromes: Mechanism, Significance, and Prognosis
 Conduction Disturbances in Acute Myocardial Infarction
Conduction Disturbances in Acute Myocardial Infarction
 Antiplatelet Therapy
Antiplatelet Therapy
 Acute Aortic Syndromes: Diagnosis and Management
Acute Aortic Syndromes: Diagnosis and Management
 Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


