Perioperative Stroke
Susanne Muehlschlegel
Laurie K. Davies
Christoph N. Seubert
CASE SUMMARY
An 82-year-old man presents for a right carotid endarterectomy. During the last 6 weeks, he had two episodes of transient weakness of the left upper extremity that lasted for approximately 2 and 14 hours, respectively. A carotid ultrasound revealed a 90% stenosis of the right internal carotid artery and an 80% stenosis of the left internal carotid artery. Computer tomographic angiography confirmed these findings and did not show significant intracranial or vertebral vascular disease.
His past medical history shows long-standing hypertension that is reasonably controlled by medications, with current blood pressures of 155/92 mmHg and 163/85 mmHg in the right and left arms, respectively. The patient had an inferior myocardial infarction 7 years before this presentation, which is evident from electrocardiogram. He denies current anginal symptoms, but his exercise tolerance is limited to approximately four metabolic equivalents (METs) because of degenerative joint disease. He has a 70-pack-year history of smoking, but quit smoking at the time of his myocardial infarction. Previous general anesthetics for an appendectomy and bilateral total hip arthroplasties more than 10 years ago were uneventful. Current medical therapy includes aspirin (acetylsalicylic acid [ASA] 81 mg), metoprolol, and a diuretic.
The patient undergoes right carotid endarterectomy under general anesthesia with shunt placement during the period of carotid cross-clamping. His intraoperative course is remarkable for a labile blood pressure, requiring the frequent administration of vasopressors and antihypertensives to control hypotension and hypertension, respectively. He awakens from anesthesia with a dense left hemiplegia, which resolves completely during the course of the next 36 hours.
What Is a Stroke and a Transient Ischemic Attack, and Why Is It Important to Know?
Stroke refers to the sudden onset of a neurologic deficit due to a focal disruption of the cerebral circulation. Stroke occurs in the setting of either brain ischemia (88% of all strokes) or hemorrhage (12%, see Fig. 24.1). Approximately 700,000 Americans have a stroke per year—that is, one person every 45 seconds. In adults, stroke is the third leading cause of death, behind heart disease and cancer, and a leading cause of serious, long-term disability.1
A transient ischemic attack (TIA) is a brief episode of neurologic dysfunction lasting <24 hours that is caused by reversible ischemia in a vascular territory. A TIA is a medical emergency, because 10% of patients with a TIA will go on to have a stroke within 30 days of the TIA, with over half of these strokes occurring within 48 hours.2 Typical TIAs can present as sudden but transient blindness in one eye (amaurosis fugax), or as other transient deficits clearly confined to an arterial territory. As TIAs have often resolved by the time the patient presents to the emergency room, leaving no neurologic deficits on examination, a detailed clinical history is critical to recognizing this sign of impending stroke. Because a TIA is considered as an impending stroke, the pathologic mechanisms, workup and treatment are the same as for ischemic stroke.
What Are the Known Risk Factors for Ischemic Stroke?
The risk factors for stroke are presented in Table 24.1. The most important risk factors are those that also
underlie atherosclerotic vascular disease in general, such as age and hypertension,1 and a history of a previous TIA or stroke (see Fig. 24.2).3 The patient, from the case description, presents with a combination of advanced age, male gender, smoking, hypertension, and evidence for generalized atherosclerosis, not unusual for stroke patients in general and for those undergoing carotid endarterectomy in particular. Less well established, but possible, risk factors for stroke are increased daytime sleepiness and obstructive sleep apnea.4
underlie atherosclerotic vascular disease in general, such as age and hypertension,1 and a history of a previous TIA or stroke (see Fig. 24.2).3 The patient, from the case description, presents with a combination of advanced age, male gender, smoking, hypertension, and evidence for generalized atherosclerosis, not unusual for stroke patients in general and for those undergoing carotid endarterectomy in particular. Less well established, but possible, risk factors for stroke are increased daytime sleepiness and obstructive sleep apnea.4
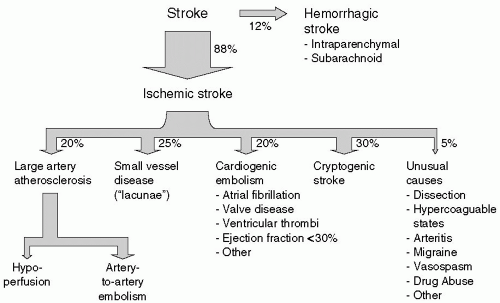 FIGURE 24.1 Etiology of stroke by mechanism, with frequency estimates. (Adapted from: Albers GW, Amarenco P, Easton JD, et al. Antithrombotic and thrombolytic therapy for ischemic stroke: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126:483S.) |
TABLE 24.1 Risk Factors for Stroke | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||
What Are the Different Types of Ischemic Stroke?
Ischemic stroke can be categorized into the following three main groups (Fig. 24.1):
Large artery atherosclerosis (with artery-to-artery embolism or occlusion)
Cardio-aortic embolism and
Small-vessel (“lacunar”) infarction
Approximately one third remain cryptogenic, meaning that no cause is found. The latest acute ischemic stroke classification is based on an evidence-based, causative system.5 The determination and classification of each stroke and TIA into these categories is essential, because stroke outcome, recurrent stroke rate, and means of secondary stroke prevention differ by stroke subtype.5 In the acute setting, clinical worsening and progression of stroke can occur within minutes or hours of presentation. Recognizing the etiology of the stroke or TIA early can potentially prevent worsening. For example, elevating the blood pressure, and therefore the cerebral perfusion pressure (CPP), in a patient with subtle right hand weakness and speech changes due to a small left middle cerebral artery (MCA) infarct could potentially prevent severe aphasia and dense right hemiparesis if
the underlying lesion is a critical (>70%) stenosis of the left internal carotid artery, with progressive hemispheric hypoperfusion. Subsequently, carotid endarterectomy would be the treatment of choice for secondary stroke prevention. Such a pattern of symptoms that may fluctuate in severity is similar to that in the introductory case description. In contrast, a lacunar stroke is most likely to improve slowly and have no acute worsening.
the underlying lesion is a critical (>70%) stenosis of the left internal carotid artery, with progressive hemispheric hypoperfusion. Subsequently, carotid endarterectomy would be the treatment of choice for secondary stroke prevention. Such a pattern of symptoms that may fluctuate in severity is similar to that in the introductory case description. In contrast, a lacunar stroke is most likely to improve slowly and have no acute worsening.
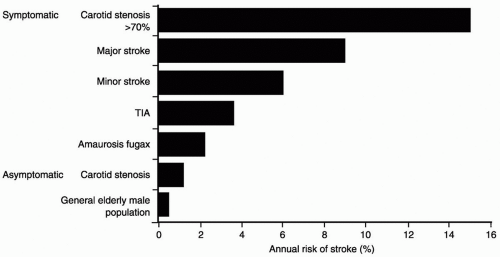 FIGURE 24.2 Annual risk of stroke among patients in various high-risk subgroups. TIA, transient ischemic attack. (Adapted from: Wilterdink JL, Easton JD. Vascular event rates in patients with atherosclerotic cerebrovascular disease. Arch Neurol. 1992;49:857.) |
How Are the Stroke Types Differentiated Clinically?
The area and vascular territory affected by ischemia can give hints to the most likely etiology. One usually starts by differentiating whether the stroke is in a single large vessel territory (e.g., MCA, anterior cerebral artery [ACA], posterior cerebral artery [PCA], or basilar artery) or affects deep penetrating arteries (“lacunae”). The clinical history and a thorough neurologic examination play a major role in this discrimination. If a patient presents with language abnormalities (aphasia) or other signs of cortical involvement (apraxia, agnosia, neglect, eye deviation, or homonymous hemianopsia, to name a few), the etiology is almost always embolic. Sometimes cortical signs are subtle and may only be revealed during examination by a neurology consultant. In rare instances, an imaging study is the only way to prove cortical involvement in either the form of a small defect in an eloquent area of the cortex that mimics lacunar symptoms or in the form of a larger defect in a noneloquent area.
What Are Lacunar (Small-Vessel) Strokes, and What Causes Them?
Weakness, sensory loss, or dysarthria (“slurred speech”) without signs of cortical involvement are most commonly seen with lacunae (“small lakes”), which encompass approximately 25% of all ischemic strokes. The name refers to the cavity or hole that remains after macrophages carry off the infarcted tissue. The size is small, the majority in the order of 2 mm3, ranging between 0.2 and 15 mm3.6 Clinicopathologic studies, largely through the efforts of C.M. Fisher, have revealed classic lacunar syndromes, such as ataxia-hemiparesis (pontine infarct),7 dysarthria-clumsy hand-syndrome (pontine or internal capsule infarct),8 pure motor hemiplegia (infarct in internal capsule or basis pontis),9 and pure-sensory stroke (infarct in ventroposterolateral thalamic nucleus, see Fig. 24.3).6 However, lacunar strokes are not limited to these syndromes. In this kind of stroke, the etiology is slowly progressive atherosclerotic disease of the deep penetrating vessels, typically from a combination of chronic hypertension, diabetes, hyperlipidemia, and smoking-causing vessel pathology such as lipohyalinosis. The most common vessels involved are the lenticulostriate arteries of the ACA and MCA, the thalamoperforators of the posterior branches of the Circle of Willis, and the paramedian branches of the basilar artery. These small perforator vessels emerge at a relatively sharp angle from a vastly larger parent vessel, which minimizes the probability that they will be entered by an embolus. As a general
rule, patients with lacunar infarcts have a relatively better clinical outcome than those with large vessel atherothrombotic strokes, but may be left with pronounced motor or cranial nerve deficits.
rule, patients with lacunar infarcts have a relatively better clinical outcome than those with large vessel atherothrombotic strokes, but may be left with pronounced motor or cranial nerve deficits.
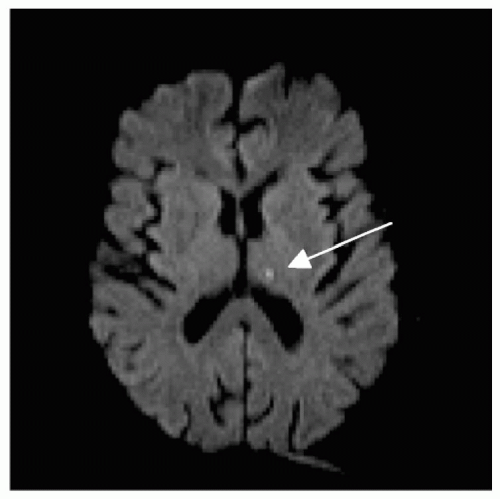 FIGURE 24.3 Diffusion-weighted MRI demonstrating an acute pure-sensory stroke from a thalamic lacunar infarction (arrow). MRI, magnetic resonance imaging. |
What Are Embolic (Large-Vessel) Strokes, and What Causes Them?
If the stroke patient displays signs of cortical involvement, thereby making embolus the most likely cause, etiologic possibilities include either atherosclerotic artery-to-artery embolus from a more proximal vessel (usually an extracranial or intracranial internal carotid or vertebral artery) or cardiac embolus (see Figs. 24.4 and 24.5). Sixty percent of all ischemic strokes are caused by cerebral embolism, but only one third have identifiable etiologies; probability is again used to determine the most likely source. If TIAs or strokes occur repeatedly on the same side, affecting either only the anterior (ACA or MCA) or posterior (PCA, basilar) circulation, the most likely etiology is from internal carotid artery or vertebral artery atherosclerosis, respectively, with distal embolization. This is based on the fact that the probability of a cardiac embolus dislodging into the same vessel each time is extremely low. However, embolization into different vessels at separate times—or even at the same times—makes the heart or aortic arch the most likely source of embolism. Multiple embolic sources have been identified as follows:10
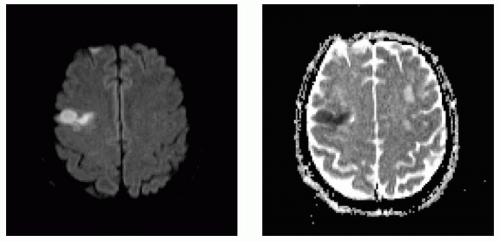 FIGURE 24.4 Acute embolic right middle cerbral artery stroke with cortical involvement, which can be seen as an abnormal signal on DWI (diffusion-weighted imaging)-MRI (bright on B1000 [left] and dark on ADC [right]). This patient suffered a cardioembolic stroke during an acute myocardial infarction. MRI, magnetic resonance imaging. ADC, apparent diffusion coefficient. |
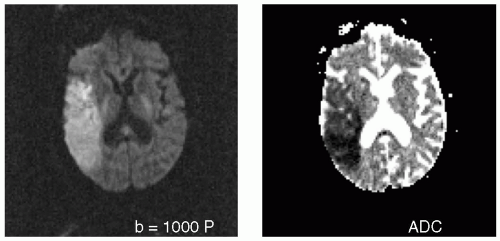 FIGURE 24.5 Large acute embolic right middle cerebral artery stroke with proximal M1 occlusion by thrombus in a patient with atrial fibrillation not on anticoagulation (B1000 on left, ADC on right). |
Atrial fibrillation
Intrinsic or mechanical valvular disease
Intracardiac thrombus (atrial or ventricular)
Atrial myxoma
Dilated cardiomyopathy
Patent foramen ovale in combination with atrial septal aneurysm
Marantic or bacterial endocarditis
Aortic arch atheromatous disease
It is very important to identify the specific source, as secondary stroke prevention differs, depending on the cause. Most cardiogenic emboli are effectively treated with coumadin anticoagulation, as was shown in the Stroke Prevention in Atrial Fibrillation Study.11 Coumadin may, however, be contraindicated in certain conditions, such as bacterial endocarditis12 or atrial myxoma,13 because emboli from these cardiac sources tend to have a high risk of hemorrhagic conversion. In addition, cerebral aneurysm formation is possible, which adds to the risk of hemorrhage in these conditions.
What Are Watershed Infarctions?
Watershed infarctions are a subgroup of atherosclerotic strokes and are caused by low-flow states in patients with severely stenosed or occluded arteries. They occur in the distal collateralization areas, the borderzones of
major cerebral arteries (ACA/MCA or MCA/PCA territory, see Fig. 24.6). A typical scenario is prolonged systemic hypotension during cardiac arrest or surgery with labile blood pressures, during which an adequate CPP cannot be maintained to perfuse the most distal endzones of the large arteries. Clinical symptoms include blood-pressure sensitive fluctuations of the neurologic examination, causing waxing and waning as stereotypic symptoms. On imaging studies, a symmetric cortical distribution is sometimes seen. The location of watershed infarctions also depends on the collateral supply of the cortex. Sources of collateral flow are derived from three areas:
major cerebral arteries (ACA/MCA or MCA/PCA territory, see Fig. 24.6). A typical scenario is prolonged systemic hypotension during cardiac arrest or surgery with labile blood pressures, during which an adequate CPP cannot be maintained to perfuse the most distal endzones of the large arteries. Clinical symptoms include blood-pressure sensitive fluctuations of the neurologic examination, causing waxing and waning as stereotypic symptoms. On imaging studies, a symmetric cortical distribution is sometimes seen. The location of watershed infarctions also depends on the collateral supply of the cortex. Sources of collateral flow are derived from three areas:
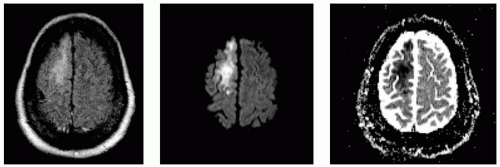 FIGURE 24.6 Middle cerebral artery/anterior cerebral artery borderzone (“watershed”) stroke after massive upper GI bleed with severe hypotension for 20 minutes or longer. FLAIR (fluid attenuated inversion recovery) image on left, DWI (diffusion-weighted imaging) with B1000 middle and DWI with ADC on right). GI, gastrointestinal. |
Collateral flow through the circle of Willis. Of note, more than one third of patients do not have a complete circle of Willis and are unable to draw on the collateral flow between the left and right hemispheres through the anterior communicating artery or between the anterior and posterior circulation through posterior communicating arteries.
Leptomeningeal collaterals that provide some overlap between endarteries within each hemisphere are another source.
The third source is extracranial to intracranial collaterals such as communications between the external carotid system and internal carotid system through the facial arteries.
What Happens When No Stroke Etiology Is Found (Cryptogenic Strokes)?
If the workup for the more common etiologies of stroke remains unrevealing, more unusual etiologies, among others, need to be considered, as follows:
Arterial dissections
Hypercoagulable states
Hyperviscosity
Central nervous system (CNS) angiitis:
Rare genetic causes (CADASIL [cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy] or MELAS [mitochondrial encephalopathy with lactacidosis and strokes]) and others
If no etiology can be found, strokes are called cryptogenic and compose approximately 30% to 40% of all strokes.14 Secondary stroke prevention strategies are difficult to determine because of the heterogeneity of this subgroup of strokes. The recent Warfarin versus Aspirin in Recurrent Stroke Study15 showed that warfarin was not superior to aspirin in the prevention of recurrent ischemic stroke in patients with a prior noncardioembolic stroke, but provided some suggestion of potential benefit in favor of oral anticoagulants in the selected subpopulation of cryptogenic stroke. The small number of patients with this subtype prevented this finding from reaching statistical significance, and therefore further studies of this subgroup are needed.
What Is the Pathophysiology and Mechanism of Acute Stroke?
Stroke and TIA are due to thrombus formation and occlusion of cerebral vessels. A thrombus (clot) tends to develop in a very limited number of locations (see Fig. 24.7). A clot may occlude a blood vessel locally at the site of its formation, or it may break off and embolize, thereby leading to occlusion of a more distal vessel, typically at a bifurcation. This is the case particularly for thrombi originating from the heart, aorta, internal carotid, or vertebral arteries.
Once the thrombus has wedged and occluded a vessel, cerebral blood flow and oxygen delivery to the downstream territory are severely decreased. If cerebral blood flow decreases below the critical limit of 18 mL/100 g/minute, irreversible neuronal damage begins to take place. The extent of neuronal damage depends on how far the blood flow decreases below this critical limit,
and how long it remains decreased. In addition to the period of time, factors such as cerebral autoregulation, CPP, the extent of cerebral collateral flow, hemoglobin concentration, and arterial oxygen saturation play crucial roles in determining the size of the infarct. CPP, hemoglobin concentration, and oxygen saturation may change significantly in the perioperative period, and therefore offer opportunities for either mitigating or aggravating secondary injury, particularly in patients with a preoperative or intraoperative stroke.
and how long it remains decreased. In addition to the period of time, factors such as cerebral autoregulation, CPP, the extent of cerebral collateral flow, hemoglobin concentration, and arterial oxygen saturation play crucial roles in determining the size of the infarct. CPP, hemoglobin concentration, and oxygen saturation may change significantly in the perioperative period, and therefore offer opportunities for either mitigating or aggravating secondary injury, particularly in patients with a preoperative or intraoperative stroke.
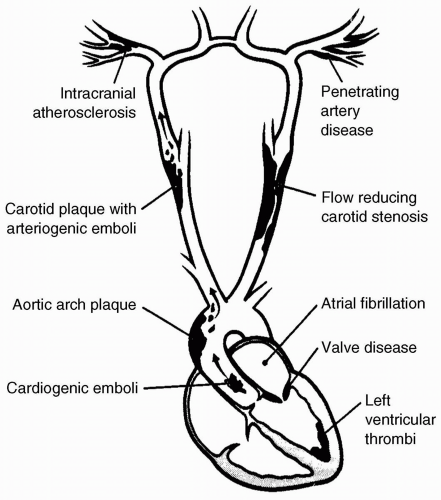 FIGURE 24.7 Common sites of thrombus formation and atheromatous disease as possible etiologies of stroke. Note that cardiogenic emboli may have different origins. (Reproduced from: Albers GW, Amarenco P, Easton JD, et al. Antithrombotic and thrombolytic therapy for ischemic stroke: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004;126:483S.) |
The tolerance for ischemia differs for different areas of the brain. It is usually greater for the brain stem than for the cortex, whereas the pyramidal neurons of the hippocampus are most vulnerable to ischemia. Tolerance for ischemia also depends on the extent of previous damage. Patients with previous strokes may have a decreased reserve for repeated ischemia. Regions in which cerebral blood flow is severely decreased will undergo infarction. These regions correspond to the core zone (center zone) of a stroke. The surrounding area of tissue around the core is referred to as the ischemic penumbra. Here, cerebral blood flow is in the range of 15 to 23 mL/100 g/minute. Brain tissue in the penumbra is at serious risk for injury but potentially salvageable (see Fig. 24.8).
In the penumbra, because of cerebral autoregulation, resistance vessels are maximally dilated, and cerebral blood flow becomes a linear function of perfusion pressure. A reduction in systemic blood pressure can, therefore, increase the size of an infarction. Speedy restoration of blood flow can save brain tissue and crucially limit the extent of a stroke. New imaging techniques such as perfusion-computer tomography or perfusion-magnetic resonance imaging are now utilized to image this “brain at risk” and may help predict stroke outcome.16,17
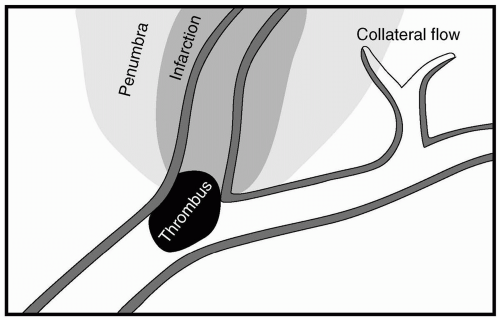 FIGURE 24.8 Ischemic stroke consists of the ischemic core and the penumbra, which is “tissue at risk.” |
What Happens at the Molecular Level in Acute Stroke?
Neuronal ischemia activates several pathologic mechanisms that, if left unchecked, not only individually lead to cell death but, in concert, augment and accelerate cell damage.18 The major pathogenic mechanisms are excitotoxicity, peri-infarct depolarizations, inflammation, and programmed cell death (apoptosis) (see Fig. 24.9).19 Their interplay and severity are dependent on the degree and duration of ischemia, and develop over time (see Fig. 24.10).
Impairment of cerebral blood flow with cessation of oxygen and glucose delivery leads to energy depletion and loss of membrane potentials, because ionic gradients can no longer be maintained. The depolarization activates presynaptic voltage-dependent Ca2+ channels, causing the massive release of glutamate and other excitatory amino acids. At the same time, energy-dependent reuptake of glutamate is impeded. There is increasing evidence that the ensuing excitotoxic damage is mediated through the activation of different glutamate receptors in the gray and white matter; activation of N-methyl-D-aspartate (NMDA) receptors predominates in damage at the neuronal level (gray matter), whereas activation of AMPA/kainate receptors is central to damage at the axonal level (white matter).20 Both lead to Na+ and Ca2+ influx, and K+ efflux, accelerating the loss of physiologic ionic gradients. Water passively follows the ionic influx. The resulting brain edema is one of the earliest markers of stroke on imaging
studies, and its extent is one of the major determinants of survival beyond the first few hours of stroke.
studies, and its extent is one of the major determinants of survival beyond the first few hours of stroke.
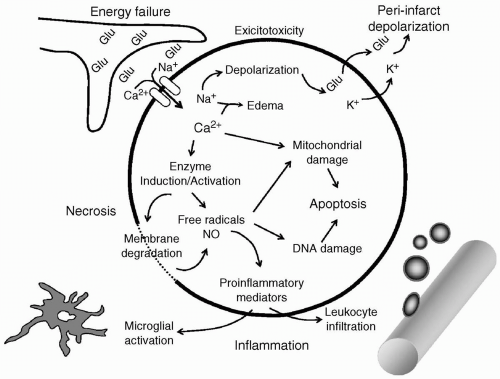 FIGURE 24.9 Simplified overview of the molecular pathophysiology in brain ischemia (see text for details). (Adapted from: Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391.) |
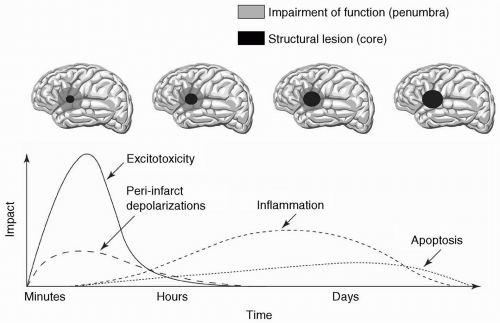 FIGURE 24.10 Cascade of damaging events in focal cerebral ischemia. The x-axis reflects the evolution of cascades over time, the y-axis shows the impact of each element of the cascade on final outcome, the schematic picture of the brain shows the extent of core and penumbra as it would be seen histologically or on imaging studies. Note the time-dependent changes in the relative importance of pathologic mechanisms at play and the time-dependent changes in the size of core and penumbra. For details, see text. (Adapted from: Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 1999;22:391.) |
The influx of Ca2+ triggers a number of nuclear and cytoplasmatic events, such as the activation of proteolytic enzymes and extracellular matrix proteins that lead to the destruction of cell structures. Ca2+ also activates phospholipase A2 and cyclooxygenase (COX), leading to free radical production, with lipid peroxidation and membrane damage. Oxygen free radicals, in turn, trigger inflammation and apoptosis. Nitric oxide (NO), which is produced by the Ca2+-dependent enzyme neuronal nitric oxide synthase (NOS), reacts with superoxide to form the highly reactive peroxinitrite, which promotes tissue damage.21 Free radicals impair mitochondrial function. The mitochondria cease to produce adenosine triphosphate (ATP), swell, and release more oxygen free-radicals22 as well as cytochrome C.23 Cytochrome C provides an important trigger for apoptosis. The neurons therefore find themselves with damage to membranes, as well as structural and functional proteins. This damage is further aggravated by a severely diminished ability to synthesize new protein for repair or survival.18
The metabolic and ionic changes as described in the preceding text affect the core and penumbra of the stroke differently. Necrosis of the core develops minutes after the onset of ischemia. Cells die rapidly through lipolysis, proteolysis, and breakdown of ionic homeostasis. The penumbra, however, can either progress to infarction more slowly by the same mechanisms as the core, or exhibit secondary phenomena, such as spreading depolarization, postischemic inflammation, reperfusion injury, and apoptosis. Peri-infarct depolarizations occur in areas where enough energy and perfusion are present to repolarize previously depolarized cells; this repolarization occurs at the expense of further energy consumption. In response to increased concentrations of glutamate and K+, the same cells depolarize again. These repetitive depolarizations have been demonstrated in animal models, occurring at a rate of several events per hour, and can be recorded for at least 6 to 8 hours after the onset of ischemia. As the number of depolarizations increase, the infarct grows larger.24
The activation of second messenger systems, increase in oxygen free radicals, and hypoxia itself trigger a number of proinflammatory genes by inducing transcription factors.19 Injured brain cells, in turn, produce mediators of inflammation, such as platelet-activating factor, TNF-α, and interleukin 1-β,19 5 to 7 days after ischemia neutrophils cross the vascular wall, followed by macrophages and monocytes. Glia cells also undergo changes. Astrocytes become hypertrophic 4 to 6 hours after ischemia, while microglia become activated. Postischemic inflammation contributes to ischemic damage by several mechanisms. Neutrophils may not only worsen ischemia by obstructing the microvasculature, but also produce toxic mediators. Examples of such mediators are NO derived in toxic amounts from inducible NOS (iNOS);25 superoxide and toxic prostanoids derived from COX 2 (2)26,27 and TNF-α, a cytokine that can exacerbate ischemic injury and trigger apoptosis.28
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


