CHAPTER 8 Pathophysiology of Acute Coronary Syndromes
Plaque Rupture and Atherothrombosis
THE ACUTE coronary syndromes (unstable angina, myocardial infarction [MI], sudden cardiac death) are a major cause of morbidity and mortality in developed countries. MI alone is the major cause of death in most Western countries.1 The rapidly increasing prevalence in developing countries, specifically South Asia and Eastern Europe, coupled with an increasing incidence of tobacco abuse, obesity, and diabetes, is predicted to make cardiovascular disease the major global cause of death by 2020.2 Atherosclerotic plaque formation within the coronary arteries with subsequent lesion disruption, platelet aggregation, and thrombus formation is the primary cause of acute coronary syndromes in humans.
During the early 1900s, the first description of the clinical presentation of acute MI was published by Obstrastzow and Straschesko.3 Shortly thereafter, Herrick4 associated the clinical presentation of acute MI with thrombotic occlusion of the coronary arteries. Much has been learned since these early observations concerning the pathophysiology of coronary artery disease and the acute coronary syndromes. This chapter reviews the pathogenesis of atherosclerosis and explores the mechanisms responsible for the sudden conversion of stable atherosclerotic plaques into unstable life-threatening atherothrombotic lesions.
Atherogenesis
Development
Atherogenesis, or the development of plaques within the walls of blood vessels, is the result of complex interactions involving blood elements, vessel wall abnormalities, and alterations in blood flow. In addition, several pathologic mechanisms play an important role, including inflammation with activation of endothelial cells and monocyte recruitment,5–9 growth with smooth muscle cell proliferation and matrix synthesis,10,11 degeneration with lipid accumulation,12,13 necrosis, calcification and ossification,14,15 and thrombosis.16 These diverse processes result in the formation of atheromatous plaques that form the substrate for future acute coronary syndromes.
Fatty Streak
Vascular injury and thrombus formation are key events in the formation and progression of atherosclerotic plaques. Fuster and colleagues17 proposed a pathophysiologic classification of vascular injury that divides the damage into three types (Fig. 8-1). Type I injury consists of functional deviations from normal endothelial function without obvious morphologic changes. Type II injury involves a deeper form of vascular damage that includes denudation of endothelial cells and intimal damage with maintenance of an intact elastic lamina. Type III injury is represented by endothelial denudation and damage to the intima and media of the vessel wall.
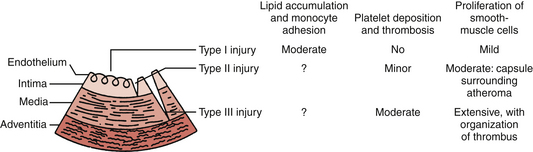
Figure 8-1 Classification of vascular injury and vascular response. See text for details.
(From Fuster V, Badimon L, Badimon J, Chesebro J: The pathogenesis of coronary artery disease and the acute coronary syndromes. N Engl J Med 1992;326:242-248.)
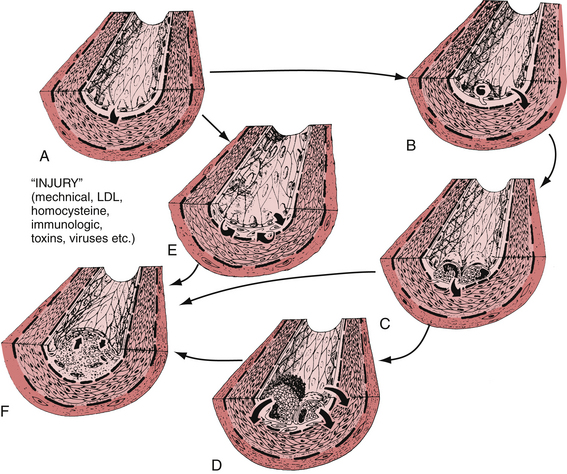
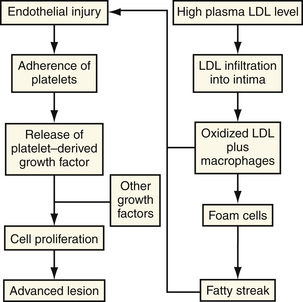
The earliest finding in spontaneous atherosclerosis is an intimal lesion containing lipid-laden macrophages and a few T lymphocytes.18 In the “response to injury hypothesis” proposed by Ross19 and others, these “fatty streaks” are thought to result from chronic injury to the arterial endothelium induced by disturbances in coronary blood flow (type I injury) and an increased vascular permeability to lipids and monocytes (Fig. 8-2). The chronic injury is primarily a disturbance in the pattern of blood flow in certain parts of the arterial tree, such as bending, branch points, or both. Certain factors, such as hypercholesterolemia, tobacco abuse, vasoactive amines, glycosylated products, infection, and immune complexes, may potentiate the effect of type I injury on the endothelium.20 This chronic, low-grade damage leads to the accumulation of lipids and macrophages at the site of injury producing the characteristic fatty streak. These lipid-laden macrophages, also known as foam cells, are derived primarily from tissue macrophages. Foam cells are formed when large amounts of intracellular lipids, mainly from modified low-density lipoprotein (LDL), are taken up via a family of macrophage scavenger receptors and internalized into the macrophage.
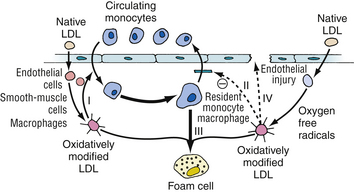
There is growing evidence that oxidized LDL is a key active component in the generation of atheroscleroses, rather than a passive substance that accumulates within macrophages. It is hypothesized that oxidized LDL has five potentially atherogenic effects: (1) monocyte chemotactic activity, (2) inhibition of macrophage migration out of the vessel wall, (3) enhanced uptake by macrophages, (4) formation of immune complexes, and (5) cytotoxicity (Fig. 8-3).
In the presence of elevated plasma LDL levels, the concentration of LDL within the intima is increased. By poorly understood mechanisms, which may involve the generation of free radicals by cellular lipoxygenases,21,22 the LDL molecule undergoes oxidative modification (peroxidation of polysaturated fatty acids) that alters its metabolism. When the LDL contains fatty acid lipid peroxides, a rapid propagation amplifies the number of free radicals and leads to extensive fragmentation of the fatty acid chains. These fragments of oxidized fatty acids attach covalently to apoprotein B.23,24 By means of specialized receptors, distinct from the LDL receptor, these modified apoprotein B molecules with the attached fatty acids are recognized by macrophages and taken up by the cell (Fig. 8-4).25 All three major cell types within the artery wall are capable of modifying LDL to a form that is recognizable by a scavenger receptor. In contrast to the LDL receptor, the scavenger LDL receptors are not downregulated in the presence of excess ligand. Cells are able to accumulate large amounts of intracellular lipid.
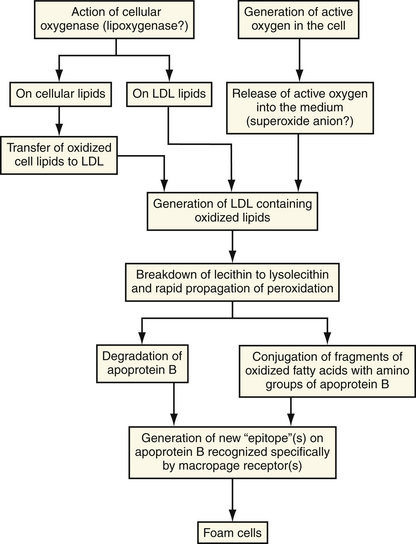
Figure 8-4 Mechanisms of oxidative modification of low-density lipoprotein (LDL) by cells.
(From Steinberg D, Parthasarathy S, Carew TE, et al: Beyond cholesterol: modification of low-density lipoprotein that increases its atherogenicity. N Engl J Med 1989;320:915-924.)
Oxidized LDL within the intima may play a role in the adhesion of circulating monocytes to the arterial wall. More recent studies have shown that oxidized LDL, but not native LDL, is a powerful chemoattractant for circulating monocytes.26 In addition, oxidized LDL is a potent inhibitor of the migration of macrophages out of the intima. By these two mechanisms, oxidized LDL may serve to attract and retain monocytes and macrophages within the vessel wall.
LDL modification by oxidation causes activation, dysfunction, apoptosis, and necrosis in human endothelial cells.27 Activated endothelial cells express leukocyte adhesion molecules, including vascular cell adhesion molecule 1 and intercellular adhesion molecule-1, causing blood cells to adhere at the sites of activation.27 Monocytes and lymphocytes preferentially adhere to these sites. A potent chemotactic agent, monocyte chemotactic protein 1, is produced by endothelial cells and smooth muscle cells. This same protein has been found in the intima of atherosclerotic lesions and in foam cells. Secretion of monocyte chemotactic protein 1 by endothelial or smooth muscle cells may be induced by oxidized LDL, suggesting a possible mechanism whereby lipids induce the recruitment of macrophages leading to the formation of early atherosclerotic lesions.28
Plaque Formation
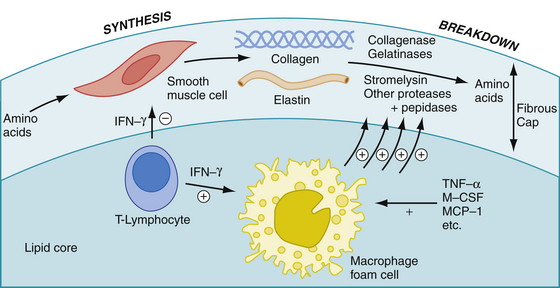
With time, fatty streaks progress into mature atherosclerotic plaques (Fig. 8-5). Macrophages recruited into the area may release toxic products that cause further damage leading to denudation of the endothelium and intimal injury (type II injury). This deeper form of injury leads to platelet adhesion. Adherent platelets, along with recruited macrophages and damaged endothelium release growth factors, such as platelet-derived growth factor (PDGF), epidermal growth factor-β, and somatomedin C. These growth factors may lead to migration and proliferation of vascular smooth muscle cells, and stimulate the production of collagen, elastin, and glycoproteins. These proteins provide the connective tissue matrix of the newly formed plaque and give it structural support. Cholesterol, derived from insudated blood lipid or extruded from dying foam cells, becomes entrapped within this matrix. The lipid and connective tissue matrix are covered by a fibromuscular cap consisting of smooth muscle cells, collagen (types I and III), and a single layer of endothelial cells. This complex constitutes the mature atherosclerotic plaque (Fig. 8-6A). Vascular smooth muscle cells synthesize and assemble the collagen fibrils, and furnish the bulk of the noncollagenous portion of the extracellular matrix of the cap. The fibromuscular cap is a dynamic structure undergoing constant remodeling through the synthesis and breakdown of essential components (Fig. 8-7; see also Plate I).
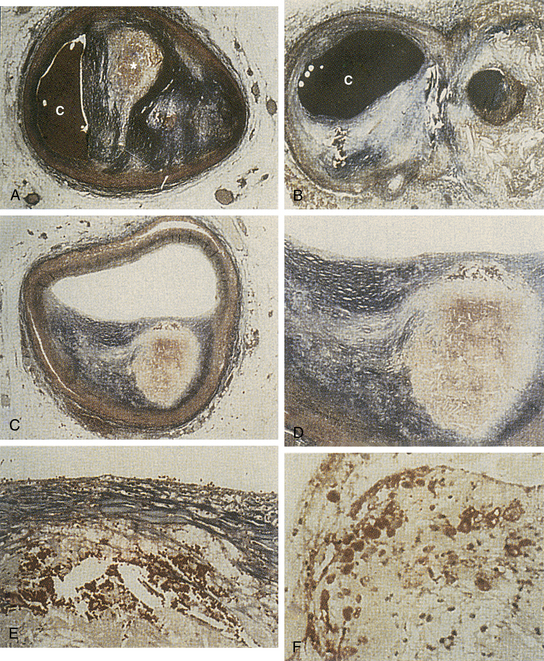
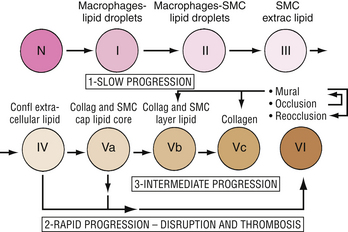
The microscopic changes that occur in spontaneous atherosclerosis have been described by Stary18 and modified by the American Heart Association (AHA).29 Using autopsy results from the coronary arteries and aortas of young people, Stary described five distinct lesions. A Stary I lesion, not apparent macroscopically, consists of isolated macrophages or foam cells within the intima of the involved vessel. These lesions are noted in 45% of infants up to 8 months of age, and eventually regress. A Stary II lesion, which is seen in adolescents, is characterized by numerous foam cells, lipid-containing smooth muscle cells, and a minimal amount of scattered extracellular lipid. Macroscopically, with Sudan IV staining, these lesions appear as a flat or raised fatty streak. In some children, more advanced lesions are noted that are characterized by an increased amount of extracellular lipid and the appearance of a raised fatty streak (Stary III) or a single confluent extracellular core (Stary IV). In adults, usually beginning in the third decade of life, two types of lesions are noted in the coronary arteries. Some plaques are mostly fibromuscular, whereas others are fibrolipid with a cap of smooth muscle cells and collagen. These latter lesions are designated as Stary V lesions.
The first three lesion types in the AHA classification are similar to those in the original Stary description. In the AHA classification, a type IV lesion has a predominance of extracellular, mostly diffuse lipid, whereas a type Va lesion has localized lipid content surrounded by a thin capsule. Additionally, type V lesions are classified further according to the amount of stenosis and fibrosis (types Vb and Vc). Type IV or Va lesions may progress slowly over time into more advanced type V lesions or undergo disruption resulting in a type VI lesion represented by a ruptured plaque with overlying thrombus (Fig. 8-8).
The composition of nonruptured atheromatous plaques is highly variable, and the factors controlling this process are poorly understood. Mature plaques consist of two components: soft, lipid-rich, atheromatous gruel and hard, collagen-rich, sclerotic tissue. The relative amounts of each component may differ with the individual plaque, but generally two populations of lesions predominate (see Fig. 8-6B). One group consists of fibrointimal lesions characterized by large amounts of fibrous tissue and relatively little atheromatous gruel. The second population consists of lipid-laden lesions with a cholesterol-rich central core and a thin outer capsule.
Differences in plaque composition have important clinical implications. Plaques causing severe stenosis tend to have a higher fibrous and lower lipid content than less stenotic lesions.30 Several studies have shown, however, the less stenotic, lipid-laden plaques to be the more clinically dangerous lesions.31 As discussed in this chapter, the soft atheromatous center may predispose the plaque to rupture, exposing the highly thrombogenic gruel and subintimal elements to blood flow, and leading to the formation of thrombus and acute myocardial ischemia in many cases.
Progression of Atherosclerosis
Atherosclerosis is a dynamic process that, without intervention, is progressive. Serial angiographic studies have shown that atherosclerotic plaques tend to enlarge over time, and that a significant number of lesions progress to total occlusion. The risk of progression to total occlusion seems to be related to the initial severity of the lesion, with more obstructive lesions progressing to total occlusion more frequently than less severe lesions.32 The factors that govern plaque progression are incompletely understood, but two mechanisms have been proposed. One mechanism involves continuation of the myointimal proliferative process produced by the chronic endothelial injury responsible for the early lesions of atherosclerosis.10 The second mechanism, which may be more important in rapidly growing plaques, involves recurrent minor fissuring of the atheromatous plaque (type Va lesion undergoing type III injury) with subsequent thrombus formation and fibrotic organization.
Chronic Endothelial Injury
In the “response-to-injury hypothesis,” progression of atherosclerotic lesions occurs via the same mechanisms responsible for the initial myointimal lesion. At least two separate processes seem to be involved. One pathway involves gross endothelial cell damage and monocyte, macrophage, and platelet recruitment with growth factor secretion leading to the formation and progression of fibrous plaques. A second pathway involves direct stimulation of endothelial cells without obvious injury. This process may increase endothelial cell turnover with increased growth factor production leading to smooth muscle cell migration and increased production of PDGF. Through these interactions, further growth of the initial fibrous plaque may occur (see Fig. 8-2).10
Recurrent Thrombosis
The second proposed mechanism of atherosclerotic progression involves recurrent minor plaque disruption with subsequent thrombus formation followed by fibrotic organization. Plaque disruption is a common event, and may be asymptomatic in many patients.33 As discussed in greater detail later, rupture of lipid-laden plaque exposes the highly thrombogenic atheromatous core and subendothelial components of the arterial wall to the circulation. This exposure results in platelet adhesion and activation with the release of growth factors and the stimulation of the coagulation cascade. This sequence of events results in the formation of thrombus, superimposed on the damaged plaque, which eventually undergoes fibrotic organization with subsequent incremental narrowing of the arterial lumen. In this model, recurrent subclinical events may lead to progressive plaque growth and, ultimately, vessel occlusion.
Several lines of evidence point to an important role of mural thrombus formation in the progression of atherosclerosis. Autopsy studies of coronary artery disease patients who died of cardiac and noncardiac causes reveal the presence of plaques with healed fissures with various stages of thrombus formation and organization and old organized coronary thrombi that are difficult to distinguish from primary atherosclerotic changes seen in the arterial wall.33–35 In situ hybridization techniques and monoclonal antibodies directed at platelets, fibrin, fibrinogen, and their degradation products reveal increased amounts of these substances within the intima, neointima, and deeper medial layer in patients with coronary artery disease.36,37 These observations suggest that products of organized thrombus are important in the growth of atherosclerotic plaques.
Platelets and mural thrombi may contribute to the progression of atherosclerosis by mechanisms other than the addition of organized fibrous layers to the plaque. As noted previously, adherent platelets are capable of secreting various mitogenic factors, including PDGF, transforming growth factor-β, and others. These factors may be involved in the proliferation, hypertrophy, and migration of smooth muscle cells, which are important steps in intimal thickening and plaque growth.17
Related posts:
 Evolution of the Coronary Care Unit: Past, Present, and Future
Evolution of the Coronary Care Unit: Past, Present, and Future
 Elevated Cardiac Troponin in the Absence of Acute Coronary Syndromes: Mechanism, Significance, and Prognosis
Elevated Cardiac Troponin in the Absence of Acute Coronary Syndromes: Mechanism, Significance, and Prognosis
 Conduction Disturbances in Acute Myocardial Infarction
Conduction Disturbances in Acute Myocardial Infarction
 Pharmacologic Interactions in the CICU
Pharmacologic Interactions in the CICU
 Acute Aortic Syndromes: Diagnosis and Management
Acute Aortic Syndromes: Diagnosis and Management
 Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


