Key Concepts
 It is important to realize that muscle relaxation does not ensure unconsciousness, amnesia, or analgesia.
It is important to realize that muscle relaxation does not ensure unconsciousness, amnesia, or analgesia.
 Depolarizing muscle relaxants act as acetylcholine (ACh) receptor agonists, whereas nondepolarizing muscle relaxants function as competitive antagonists.
Depolarizing muscle relaxants act as acetylcholine (ACh) receptor agonists, whereas nondepolarizing muscle relaxants function as competitive antagonists.
 Because depolarizing muscle relaxants are not metabolized by acetylcholinesterase, they diffuse away from the neuromuscular junction and are hydrolyzed in the plasma and liver by another enzyme, pseudocholinesterase (nonspecific cholinesterase, plasma cholinesterase, or butyrylcholinesterase).
Because depolarizing muscle relaxants are not metabolized by acetylcholinesterase, they diffuse away from the neuromuscular junction and are hydrolyzed in the plasma and liver by another enzyme, pseudocholinesterase (nonspecific cholinesterase, plasma cholinesterase, or butyrylcholinesterase).
 With the exception of mivacurium, nondepolarizing agents are not significantly metabolized by either acetylcholinesterase or pseudocholinesterase. Reversal of their blockade depends on redistribution, gradual metabolism, and excretion of the relaxant by the body, or administration of specific reversal agents (eg, cholinesterase inhibitors) that inhibit acetylcholinesterase enzyme activity.
With the exception of mivacurium, nondepolarizing agents are not significantly metabolized by either acetylcholinesterase or pseudocholinesterase. Reversal of their blockade depends on redistribution, gradual metabolism, and excretion of the relaxant by the body, or administration of specific reversal agents (eg, cholinesterase inhibitors) that inhibit acetylcholinesterase enzyme activity.
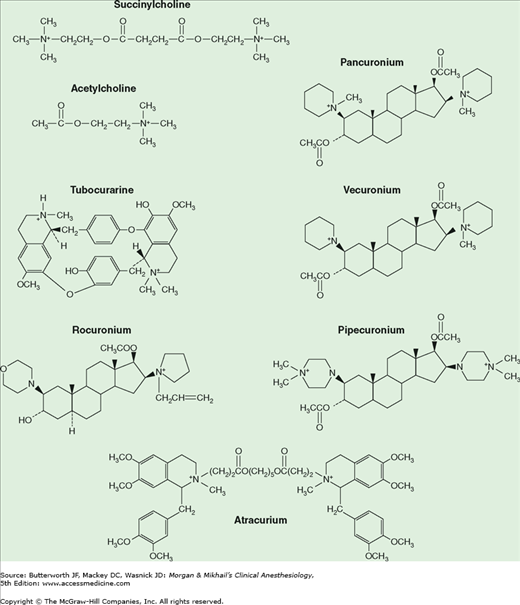
 Muscle relaxants owe their paralytic properties to mimicry of ACh. For example, succinylcholine consists of two joined ACh molecules.
Muscle relaxants owe their paralytic properties to mimicry of ACh. For example, succinylcholine consists of two joined ACh molecules.
 Compared with patients with low enzyme levels or heterozygous atypical enzyme in whom blockade duration is doubled or tripled, patients with homozygous atypical enzyme will have a very long blockade (eg, 4-8 h) following succinylcholine administration.
Compared with patients with low enzyme levels or heterozygous atypical enzyme in whom blockade duration is doubled or tripled, patients with homozygous atypical enzyme will have a very long blockade (eg, 4-8 h) following succinylcholine administration.
 Succinylcholine is considered contraindicated in the routine management of children and adolescents because of the risk of hyperkalemia, rhabdomyolysis, and cardiac arrest in children with undiagnosed myopathies.
Succinylcholine is considered contraindicated in the routine management of children and adolescents because of the risk of hyperkalemia, rhabdomyolysis, and cardiac arrest in children with undiagnosed myopathies.
 Normal muscle releases enough potassium during succinylcholine-induced depolarization to raise serum potassium by 0.5 mEq/L. Although this is usually insignificant in patients with normal baseline potassium levels, a life-threatening potassium elevation is possible in patients with burn injury, massive trauma, neurological disorders, and several other conditions.
Normal muscle releases enough potassium during succinylcholine-induced depolarization to raise serum potassium by 0.5 mEq/L. Although this is usually insignificant in patients with normal baseline potassium levels, a life-threatening potassium elevation is possible in patients with burn injury, massive trauma, neurological disorders, and several other conditions.
 Doxacurium, pancuronium, vecuronium, and pipecuronium are partially excreted by the kidneys, and their action is prolonged in patients with renal failure.
Doxacurium, pancuronium, vecuronium, and pipecuronium are partially excreted by the kidneys, and their action is prolonged in patients with renal failure.
 Cirrhotic liver disease and chronic renal failure often result in an increased volume of distribution and a lower plasma concentration for a given dose of water-soluble drugs, such as muscle relaxants. On the other hand, drugs dependent on hepatic or renal excretion may demonstrate prolonged clearance. Thus, depending on the drug, a greater initial dose—but smaller maintenance doses—might be required in these diseases.
Cirrhotic liver disease and chronic renal failure often result in an increased volume of distribution and a lower plasma concentration for a given dose of water-soluble drugs, such as muscle relaxants. On the other hand, drugs dependent on hepatic or renal excretion may demonstrate prolonged clearance. Thus, depending on the drug, a greater initial dose—but smaller maintenance doses—might be required in these diseases.
 Atracurium and cisatracurium undergo degradation in plasma at physiological pH and temperature by organ-independent Hofmann elimination. The resulting metabolites (a monoquaternary acrylate and laudanosine) have no intrinsic neuromuscular blocking effects.
Atracurium and cisatracurium undergo degradation in plasma at physiological pH and temperature by organ-independent Hofmann elimination. The resulting metabolites (a monoquaternary acrylate and laudanosine) have no intrinsic neuromuscular blocking effects.
 Hypertension and tachycardia may occur in patients given pancuronium. These cardiovascular effects are caused by the combination of vagal blockade and catecholamine release from adrenergic nerve endings.
Hypertension and tachycardia may occur in patients given pancuronium. These cardiovascular effects are caused by the combination of vagal blockade and catecholamine release from adrenergic nerve endings.
 Long-term administration of vecuronium to patients in intensive care units has resulted in prolonged neuromuscular blockade (up to several days), possibly from accumulation of its active 3-hydroxy metabolite, changing drug clearance, or the development of a polyneuropathy.
Long-term administration of vecuronium to patients in intensive care units has resulted in prolonged neuromuscular blockade (up to several days), possibly from accumulation of its active 3-hydroxy metabolite, changing drug clearance, or the development of a polyneuropathy.
 Rocuronium (0.9-1.2 mg/kg) has an onset of action that approaches succinylcholine (60-90 s), making it a suitable alternative for rapid-sequence inductions, but at the cost of a much longer duration of action.
Rocuronium (0.9-1.2 mg/kg) has an onset of action that approaches succinylcholine (60-90 s), making it a suitable alternative for rapid-sequence inductions, but at the cost of a much longer duration of action.
Neuromuscular Blocking Agents: Introduction
Skeletal muscle relaxation can be produced by deep inhalational anesthesia, regional nerve block, or neuromuscular blocking agents (commonly called muscle relaxants). In 1942, Harold Griffith published the results of a study using an extract of curare (a South American arrow poison) during anesthesia. Following the introduction of succinylcholine as a “new approach to muscular relaxation,” these agents rapidly became a routine part of the anesthesiologist’s drug arsenal. However, as noted by Beecher and Todd in 1954: “[m]uscle relaxants given inappropriately may provide the surgeon with optimal [operating] conditions in… a patient [who] is paralyzed but not anesthetized—a state [that] is wholly unacceptable for the patient.”  In other words, muscle relaxation does not ensure unconsciousness, amnesia, or analgesia. This chapter reviews the principles of neuromuscular transmission and presents the mechanisms of action, physical structures, routes of elimination, recommended dosages, and side effects of several muscle relaxants.
In other words, muscle relaxation does not ensure unconsciousness, amnesia, or analgesia. This chapter reviews the principles of neuromuscular transmission and presents the mechanisms of action, physical structures, routes of elimination, recommended dosages, and side effects of several muscle relaxants.
Neuromuscular Transmission
Association between a motor neuron and a muscle cell occurs at the neuromuscular junction (Figure 11-1). The cell membranes of the neuron and muscle fiber are separated by a narrow (20-nm) gap, the synaptic cleft. As a nerve’s action potential depolarizes its terminal, an influx of calcium ions through voltage-gated calcium channels into the nerve cytoplasm allows storage vesicles to fuse with the terminal plasma membrane and release their contents (acetylcholine [ACh]). The ACh molecules diffuse across the synaptic cleft to bind with nicotinic cholinergic receptors on a specialized portion of the muscle membrane, the motor end-plate. Each neuromuscular junction contains approximately 5 million of these receptors, but activation of only about 500,000 receptors is required for normal muscle contraction.
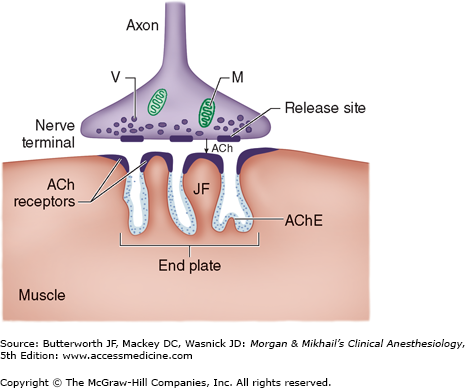
The structure of ACh receptors varies in different tissues and at different times in development. Each ACh receptor in the neuromuscular junction normally consists of five protein subunits; two α subunits; and single β, δ, and ϵ subunits. Only the two identical α subunits are capable of binding ACh molecules. If both binding sites are occupied by ACh, a conformational change in the subunits briefly (1 ms) opens an ion channel in the core of the receptor (Figure 11-2). The channel will not open if ACh binds on only one site. In contrast to the normal (or mature) junctional ACh receptor, another isoform contains a γ subunit instead of the ϵ subunit. This isoform is referred to as the fetal or immature receptor because it is in the form initially expressed in fetal muscle. It is also often referred to as extrajunctional because, unlike the mature isoform, it may be located anywhere in the muscle membrane, inside or outside the neuromuscular junction when expressed in adults.
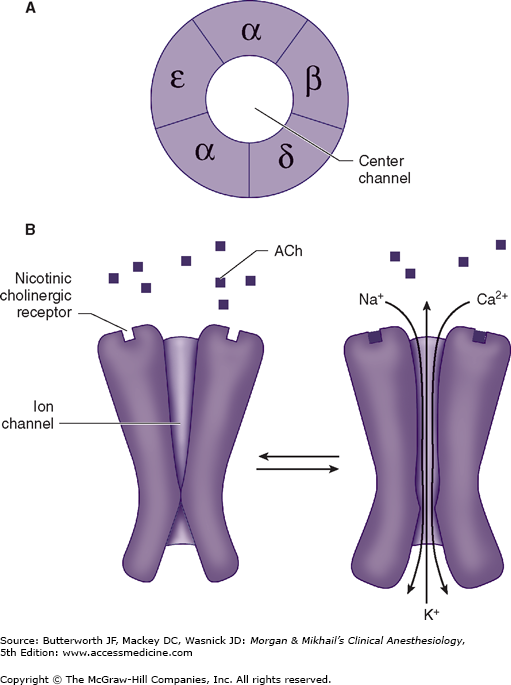
Cations flow through the open ACh receptor channel (sodium and calcium in; potassium out), generating an end-plate potential. The contents of a single vesicle, a quantum of ACh (104 molecules per quantum), produce a miniature end-plate potential. The number of quanta released by each nerve impulse, normally at least 200, is very sensitive to extracellular ionized calcium concentration; increasing calcium concentration increases the number of quanta released. When enough receptors are occupied by ACh, the end-plate potential will be sufficiently strong to depolarize the perijunctional membrane. Voltage-gated sodium channels within this portion of the muscle membrane open when a threshold voltage is developed across them, as opposed to end-plate receptors that open when ACh is applied (Figure 11-3). Perijunctional areas of muscle membrane have a higher density of these sodium channels than other parts of the membrane. The resulting action potential propagates along the muscle membrane and T-tubule system, opening sodium channels and releasing calcium from the sarcoplasmic reticulum. This intracellular calcium allows the contractile proteins actin and myosin to interact, bringing about muscle contraction. The amount of ACh released and the number of receptors subsequently activated will normally far exceed the minimum required for the initiation of an action potential. The nearly 10-fold margin of safety is lost in Eaton-Lambert myasthenic syndrome (decreased release of ACh) and myasthenia gravis (decreased number of receptors).
Figure 11-3
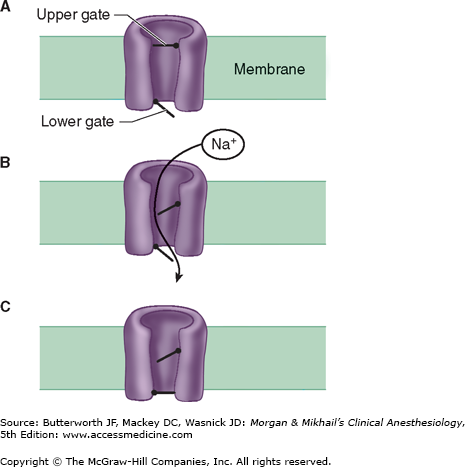
Schematic of the sodium channel. The sodium channel is a transmembrane protein that can be conceptualized as having two gates. Sodium ions pass only when both gates are open. Opening of the gates is time dependent and voltage dependent. The channel therefore possesses three functional states. At rest, the lower gate is open but the upper gate is closed (A). When the muscle membrane reaches threshold voltage depolarization, the upper gate opens and sodium can pass (B). Shortly after the upper gate opens the time-dependent lower gate closes (C). When the membrane repolarizes to its resting voltage, the upper gate closes and the lower gate opens (A).
ACh is rapidly hydrolyzed into acetate and choline by the substrate-specific enzyme acetylcholinesterase. This enzyme (also called specific cholinesterase or true cholinesterase) is embedded into the motor end-plate membrane immediately adjacent to the ACh receptors. After unbinding ACh, the receptors’ ion channels close, permitting the end-plate to repolarize. Calcium is resequestered in the sarcoplasmic reticulum, and the muscle cell relaxes.
Distinctions between Depolarizing & Nondepolarizing Blockade
Neuromuscular blocking agents are divided into two classes: depolarizing and nondepolarizing (Table 11-1). This division reflects distinct differences in the mechanism of action, response to peripheral nerve stimulation, and reversal of block.
Similar to ACh, all neuromuscular blocking agents are quaternary ammonium compounds whose positively charged nitrogen imparts an affinity to nicotinic ACh receptors. Whereas most agents have two quaternary ammonium atoms, a few have one quaternary ammonium cation and one tertiary amine that is protonated at physiological pH.
Depolarizing muscle relaxants very closely resemble ACh and readily bind to ACh receptors, generating a muscle action potential. Unlike ACh, however, these drugs are not metabolized by acetylcholinesterase, and their concentration in the synaptic cleft does not fall as rapidly, resulting in a prolonged depolarization of the muscle end-plate.
Continuous end-plate depolarization causes muscle relaxation because opening of perijunctional sodium channels is time limited (sodium channels rapidly “inactivate” with continuing depolarization) (Figure 11-3). After the initial excitation and opening (Figure 11-3B), these sodium channels inactivate (Figure 11-3C) and cannot reopen until the end-plate repolarizes. The end-plate cannot repolarize as long as the depolarizing muscle relaxant continues to bind to ACh receptors; this is called a phase I block. After a period of time, prolonged end-plate depolarization can cause poorly understood changes in the ACh receptor that result in a phase II block, which clinically resembles that of nondepolarizing muscle relaxants.
Nondepolarizing muscle relaxants bind ACh receptors but are incapable of inducing the conformational change necessary for ion channel opening. Because ACh is prevented from binding to its receptors, no end-plate potential develops. Neuromuscular blockade occurs even if only one α subunit is blocked.
 Thus, depolarizing muscle relaxants act as ACh receptor agonists, whereas nondepolarizing muscle relaxants function as competitive antagonists. This basic difference in mechanism of action explains their varying effects in certain disease states. For example, conditions associated with a chronic decrease in ACh release (eg, muscle denervation injuries) stimulate a compensatory increase in the number of ACh receptors within muscle membranes. These states also promote the expression of the immature (extrajunctional) isoform of the ACh receptor, which displays low channel conductance properties and prolonged open-channel time. This up-regulation causes an exaggerated response to depolarizing muscle relaxants (with more receptors being depolarized), but a resistance to nondepolarizing relaxants (more receptors that must be blocked). In contrast, conditions associated with fewer ACh receptors (eg, down-regulation in myasthenia gravis) demonstrate a resistance to depolarizing relaxants and an increased sensitivity to nondepolarizing relaxants.
Thus, depolarizing muscle relaxants act as ACh receptor agonists, whereas nondepolarizing muscle relaxants function as competitive antagonists. This basic difference in mechanism of action explains their varying effects in certain disease states. For example, conditions associated with a chronic decrease in ACh release (eg, muscle denervation injuries) stimulate a compensatory increase in the number of ACh receptors within muscle membranes. These states also promote the expression of the immature (extrajunctional) isoform of the ACh receptor, which displays low channel conductance properties and prolonged open-channel time. This up-regulation causes an exaggerated response to depolarizing muscle relaxants (with more receptors being depolarized), but a resistance to nondepolarizing relaxants (more receptors that must be blocked). In contrast, conditions associated with fewer ACh receptors (eg, down-regulation in myasthenia gravis) demonstrate a resistance to depolarizing relaxants and an increased sensitivity to nondepolarizing relaxants.
Some drugs may interfere with the function of the ACh receptor without acting as an agonist or antagonist. They interfere with normal functioning of the ACh receptor binding site or with the opening and closing of the receptor channel. These may include inhaled anesthetic agents, local anesthetics, and ketamine. The ACh receptor-lipid membrane interface may be an important site of action.
Drugs may also cause either closed or open channel blockade. During closed channel blockade, the drug physically plugs up the channel, preventing passage of cations whether or not ACh has activated the receptor. Open channel blockade is use dependent, because the drug enters and obstructs the ACh receptor channel only after it is opened by ACh binding. The clinical relevance of open channel blockade is unknown. Based on laboratory experiments, one would expect that increasing the concentration of ACh with a cholinesterase inhibitor would not overcome this form of neuromuscular blockade. Drugs that may cause channel block in the laboratory include neostigmine, some antibiotics, cocaine, and quinidine. Other drugs may impair the presynaptic release of ACh. Prejunctional receptors play a role in mobilizing ACh to maintain muscle contraction. Blocking these receptors can lead to a fading of the train-of-four response.
 Because succinylcholine is not metabolized by acetylcholinesterase, it unbinds the receptor and diffuses away from the neuromuscular junction to be hydrolyzed in the plasma and liver by another enzyme, pseudocholinesterase (nonspecific cholinesterase, plasma cholinesterase, or butyrylcholinesterase). Fortunately, this is a fairly rapid process, because no specific agent to reverse a depolarizing blockade is available.
Because succinylcholine is not metabolized by acetylcholinesterase, it unbinds the receptor and diffuses away from the neuromuscular junction to be hydrolyzed in the plasma and liver by another enzyme, pseudocholinesterase (nonspecific cholinesterase, plasma cholinesterase, or butyrylcholinesterase). Fortunately, this is a fairly rapid process, because no specific agent to reverse a depolarizing blockade is available.
 With the exception of the discontinued drug mivacurium, nondepolarizing agents are not metabolized by either acetylcholinesterase or pseudocholinesterase. Reversal of their blockade depends on unbinding the receptor, redistribution, metabolism, and excretion of the relaxant by the body, or administration of specific reversal agents (eg, cholinesterase inhibitors) that inhibit acetylcholinesterase enzyme activity. Because this inhibition increases the amount of ACh that is available at the neuromuscular junction and can compete with the nondepolarizing agent, clearly, the reversal agents are of no benefit in reversing a depolarizing block. In fact, by increasing neuromuscular junction ACh concentration and inhibiting pseudocholinesterase-induced metabolism of succinylcholine, cholinesterase inhibitors can prolong neuromuscular blockade produced by succinylcholine. The ONLY time neostigmine reverses neuromuscular block after succinylcholine is when there is a phase II block (fade of the train-of-four) AND sufficient time has passed for the circulating concentration of succinylcholine to be negligible.
With the exception of the discontinued drug mivacurium, nondepolarizing agents are not metabolized by either acetylcholinesterase or pseudocholinesterase. Reversal of their blockade depends on unbinding the receptor, redistribution, metabolism, and excretion of the relaxant by the body, or administration of specific reversal agents (eg, cholinesterase inhibitors) that inhibit acetylcholinesterase enzyme activity. Because this inhibition increases the amount of ACh that is available at the neuromuscular junction and can compete with the nondepolarizing agent, clearly, the reversal agents are of no benefit in reversing a depolarizing block. In fact, by increasing neuromuscular junction ACh concentration and inhibiting pseudocholinesterase-induced metabolism of succinylcholine, cholinesterase inhibitors can prolong neuromuscular blockade produced by succinylcholine. The ONLY time neostigmine reverses neuromuscular block after succinylcholine is when there is a phase II block (fade of the train-of-four) AND sufficient time has passed for the circulating concentration of succinylcholine to be negligible.
Sugammadex, a cyclodextrin, is the first selective relaxant-binding agent; it exerts its reversal effect by forming tight complexes in a 1:1 ratio with steroidal nondepolarizing agents (vecuronium, rocuronium,). This drug has been in use in the European Union for the past few years, but is not yet commercially available in the United States.
The newer neuromuscular blocking agents, such as gantacurium, which are still under investigation, show promise as ultrashort-acting nondepolarizing agents; they undergo chemical degradation by rapid adduction with L-cysteine.
The use of peripheral nerve stimulators to monitor neuromuscular function is discussed in Chapter 6. Four patterns of electrical stimulation with supramaximal square-wave pulses are considered:
Tetany: A sustained stimulus of 50-100 Hz, usually lasting 5 sec.
Single twitch: A single pulse 0.2 ms in duration.
Train-of-four: A series of four twitches in 2 s (2-Hz frequency), each 0.2 ms long.
Double-burst stimulation (DBS): Three short (0.2 ms) high-frequency stimulations separated by a 20-ms interval (50 Hz) and followed 750 ms later by two (DBS3,2) or three (DBS3,3) additional impulses.
The occurrence of fade, a gradual diminution of evoked response during prolonged or repeated nerve stimulation, is indicative of a nondepolarizing block (Table 11-2), or of a phase II block if only succinylcholine has been administered. Fade may be due to a prejunctional effect of nondepolarizing relaxants that reduces the amount of ACh in the nerve terminal available for release during stimulation (blockade of ACh mobilization). Adequate clinical recovery correlates well with the absence of fade. Because fade is more obvious during sustained tetanic stimulation or double-burst stimulation than following a train-of-four pattern or repeated twitches, the first two patterns are the preferred methods for determining adequacy of recovery from a nondepolarizing block.
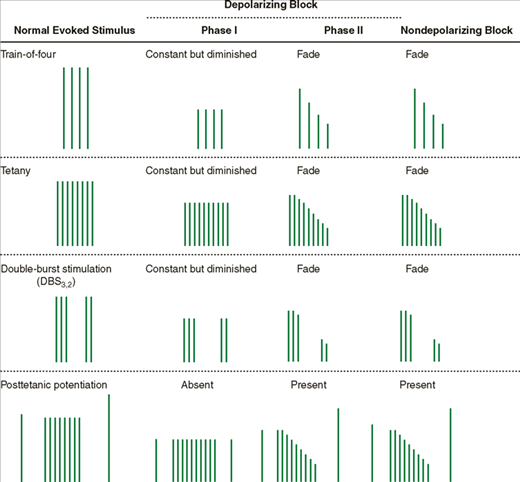
The ability of tetanic stimulation during a partial nondepolarizing block to increase the evoked response to a subsequent twitch is termed posttetanic potentiation. This phenomenon may relate to a transient increase in ACh mobilization following tetanic stimulation.
In contrast, a phase I depolarization block from succinylcholine does not exhibit fade during tetanus or train-of-four; neither does it demonstrate posttetanic potentiation. With longer infusions of succinylcholine, however, the quality of the block will sometimes change to resemble a nondepolarizing block (phase II block).
Newer quantitative methods of assessment of neuromuscular blockade, such as acceleromyography, permit determination of exact train-of-four ratios, as opposed to subjective interpretations. Acceleromyography may reduce the incidence of unexpected postoperative residual neuromuscular blockade.
Depolarizing Muscle Relaxants
The only depolarizing muscle relaxant in clinical use today is succinylcholine.
 Succinylcholine—also called diacetylcholine or suxamethonium—consists of two joined ACh molecules (Figure 11-4). This structure underlies succinylcholine’s mechanism of action, side effects, and metabolism.
Succinylcholine—also called diacetylcholine or suxamethonium—consists of two joined ACh molecules (Figure 11-4). This structure underlies succinylcholine’s mechanism of action, side effects, and metabolism.
The popularity of succinylcholine is due to its rapid onset of action (30-60 s) and short duration of action (typically less than 10 min). Its rapid onset of action relative to other neuromuscular blockers is largely due to the relative overdose that is usually administered. Succinylcholine, like all neuromuscular blockers, has a small volume of distribution due to its very low lipid solubility, and this also underlies a rapid onset of action. As succinylcholine enters the circulation, most of it is rapidly metabolized by pseudocholinesterase into succinylmonocholine. This process is so efficient that only a small fraction of the injected dose ever reaches the neuromuscular junction. As drug levels fall in blood, succinylcholine molecules diffuse away from the neuromuscular junction, limiting the duration of action. However, this duration of action can be prolonged by high doses, infusion of succinylcholine, or abnormal metabolism. The latter may result from hypothermia, reduced pseudocholinesterase levels, or a genetically aberrant enzyme. Hypothermia decreases the rate of hydrolysis. Reduced levels of pseudocholinesterase (measured as units per liter) accompany pregnancy, liver disease, renal failure, and certain drug therapies (Table 11-3). Reduced pseudocholinesterase levels generally produce only modest prolongation of succinylcholine’s actions (2-20 min).
| Drug | Description |
|---|---|
| Echothiophate | Organophosphate use for glaucoma |
Neostigmine Pyridostigmine | Cholinesterase inhibitors |
| Phenelzine | Monoamine oxidase inhibitor |
| Cyclophosphamide | Antineoplastic agent |
| Metoclopramide | Antiemetic/prokinetic agent |
| Esmolol | β-Blocker |
| Pancuronium | Nondepolarizing muscle relaxant |
| Oral contraceptives | Various agents |
One in 25-30 patients of European extraction is a heterozygote with one normal and one abnormal (atypical) pseudocholinesterase gene, resulting in a slightly prolonged block (20-30 min). Even fewer (1 in 3000) patients have two copies of the most prevalent abnormal gene (homozygous atypical) that produce an enzyme with little or no affinity for succinylcholine.  In contrast to the doubling or tripling of blockade duration seen in patients with low enzyme levels or heterozygous atypical enzyme, patients with homozygous atypical enzyme will have a very long blockade (eg, 4-8 h) following administration of succinylcholine. Of the recognized abnormal pseudocholinesterase genes, the dibucaine-resistant (variant) allele, which produces an enzyme with 1/100 of normal affinity for succinylcholine, is the most common. Other variants include fluoride-resistant and silent (no activity) alleles.
In contrast to the doubling or tripling of blockade duration seen in patients with low enzyme levels or heterozygous atypical enzyme, patients with homozygous atypical enzyme will have a very long blockade (eg, 4-8 h) following administration of succinylcholine. Of the recognized abnormal pseudocholinesterase genes, the dibucaine-resistant (variant) allele, which produces an enzyme with 1/100 of normal affinity for succinylcholine, is the most common. Other variants include fluoride-resistant and silent (no activity) alleles.
Dibucaine, a local anesthetic, inhibits normal pseudocholinesterase activity by 80%, but inhibits atypical enzyme activity by only 20%. Serum from an individual who is heterozygous for the atypical enzyme is characterized by an intermediate 40% to 60% inhibition. The percentage of inhibition of pseudocholinesterase activity is termed the dibucaine number. A patient with normal pseudocholinesterase has a dibucaine number of 80; a homozygote for the most common abnormal allele will have a dibucaine number of 20. The dibucaine number measures pseudocholinesterase function, not the amount of enzyme. Therefore, adequacy of pseudocholinesterase can be determined in the laboratory quantitatively in units per liter (a minor factor) and qualitatively by dibucaine number (the major factor). Prolonged paralysis from succinylcholine caused by abnormal pseudocholinesterase (atypical cholinesterase) should be treated with continued mechanical ventilation and sedation until muscle function returns to normal by clinical signs. Such unsedated patients do NOT appreciate unnecessary, repetitive use of nerve stimulation when all members of a department come by to confirm the diagnosis.
The effects of muscle relaxants can be modified by concurrent drug therapy (Table 11-4). Succinylcholine is involved in two interactions deserving special comment.
| Drug | Effect on Depolarizing Blockade | Effect on Nondepolarizing Blockade | Comments |
|---|---|---|---|
| Antibiotics | + | + | Streptomycin, aminoglycosides, kanamycin, neomycin, colistin, polymyxin, tetracycline, lincomycin, clindamycin |
| Anticonvulsants | ? | − | Phenytoin, carbamazepine, primidone, sodium valproate |
| Antiarrhythmics | + | + | Quinidine, calcium channel blockers |
| Cholinesterase inhibitors | + | − | Neostigmine, pyridostigmine |
| Dantrolene | ? | + | Used in treatment of malignant hyperthermia (has quaternary ammonium group) |
| Inhalational anesthetics | + | + | Volatile anesthetics |
| Ketamine | ? | + | |
| Local anesthetics | + | + | High doses only |
| Lithium carbonate | + | ? | Prolongs onset and duration of succinylcholine |
| Magnesium sulfate | + | + | Doses used to treat preeclampsia and eclampsia of pregnancy |
Although cholinesterase inhibitors reverse nondepolarizing paralysis, they markedly prolong a depolarizing phase I block by two mechanisms. By inhibiting acetylcholinesterase, they lead to a higher ACh concentration at the nerve terminal, which intensifies depolarization. They also reduce the hydrolysis of succinylcholine by inhibiting pseudocholinesterase. Organophosphate pesticides, for example, cause an irreversible inhibition of acetylcholinesterase and can prolong the action of succinylcholine by 20-30 min. Echothiophate eye drops, used in the past for glaucoma, can markedly prolong succinylcholine by this mechanism.
In general, small doses of nondepolarizing relaxants antagonize a depolarizing phase I block. Because the drugs occupy some ACh receptors, depolarization by succinylcholine is partially prevented.
If enough depolarizing agent is administered to develop a phase II block, a nondepolarizer will potentiate paralysis.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


