Key Concepts
 The greater the uptake of anesthetic agent, the greater the difference between inspired and alveolar concentrations, and the slower the rate of induction.
The greater the uptake of anesthetic agent, the greater the difference between inspired and alveolar concentrations, and the slower the rate of induction.
 Three factors affect anesthetic uptake: solubility in the blood, alveolar blood flow, and the difference in partial pressure between alveolar gas and venous blood.
Three factors affect anesthetic uptake: solubility in the blood, alveolar blood flow, and the difference in partial pressure between alveolar gas and venous blood.
 Low-output states predispose patients to overdosage with soluble agents, as the rate of rise in alveolar concentrations will be markedly increased.
Low-output states predispose patients to overdosage with soluble agents, as the rate of rise in alveolar concentrations will be markedly increased.
 Many of the factors that speed induction also speed recovery: elimination of rebreathing, high fresh gas flows, low anesthetic-circuit volume, low absorption by the anesthetic circuit, decreased solubility, high cerebral blood flow, and increased ventilation.
Many of the factors that speed induction also speed recovery: elimination of rebreathing, high fresh gas flows, low anesthetic-circuit volume, low absorption by the anesthetic circuit, decreased solubility, high cerebral blood flow, and increased ventilation.
 The unitary hypothesis proposes that all inhalation agents share a common mechanism of action at the molecular level. This is supported by the observation that the anesthetic potency of inhalation agents correlates directly with their lipid solubility (Meyer-Overton rule). There is an ongoing debate as to the mechanism of anesthetic action. Anesthetic interactions at specific protein ion channels, as well as more nonspecific membrane effects, may combine to produce the anesthetized state.
The unitary hypothesis proposes that all inhalation agents share a common mechanism of action at the molecular level. This is supported by the observation that the anesthetic potency of inhalation agents correlates directly with their lipid solubility (Meyer-Overton rule). There is an ongoing debate as to the mechanism of anesthetic action. Anesthetic interactions at specific protein ion channels, as well as more nonspecific membrane effects, may combine to produce the anesthetized state.
 The minimum alveolar concentration (MAC) is the alveolar concentration of an inhaled anesthetic that prevents movement in 50% of patients in response to a standardized stimulus (eg, surgical incision).
The minimum alveolar concentration (MAC) is the alveolar concentration of an inhaled anesthetic that prevents movement in 50% of patients in response to a standardized stimulus (eg, surgical incision).
 Prolonged exposure to anesthetic concentrations of nitrous oxide can result in bone marrow depression (megaloblastic anemia) and even neurological deficiencies (peripheral neuropathies).
Prolonged exposure to anesthetic concentrations of nitrous oxide can result in bone marrow depression (megaloblastic anemia) and even neurological deficiencies (peripheral neuropathies).
 Halothane hepatitis is extremely rare (1 per 35,000 cases). Patients exposed to multiple halothane anesthetics at short intervals, middle-aged obese women, and persons with a familial predisposition to halothane toxicity or a personal history of toxicity are considered to be at increased risk. Desflurane and isoflurane undergo much less metabolism than halothane, resulting in fewer of the metabolite protein adducts that lead to immunologically mediated hepatic injury.
Halothane hepatitis is extremely rare (1 per 35,000 cases). Patients exposed to multiple halothane anesthetics at short intervals, middle-aged obese women, and persons with a familial predisposition to halothane toxicity or a personal history of toxicity are considered to be at increased risk. Desflurane and isoflurane undergo much less metabolism than halothane, resulting in fewer of the metabolite protein adducts that lead to immunologically mediated hepatic injury.
 Isoflurane dilates coronary arteries, but is not nearly as potent a dilator as nitroglycerin or adenosine. Dilation of normal coronary arteries could theoretically divert blood away from fixed stenotic lesions.
Isoflurane dilates coronary arteries, but is not nearly as potent a dilator as nitroglycerin or adenosine. Dilation of normal coronary arteries could theoretically divert blood away from fixed stenotic lesions.
 The low solubility of desflurane in blood and body tissues causes a very rapid induction of and emergence from anesthesia.
The low solubility of desflurane in blood and body tissues causes a very rapid induction of and emergence from anesthesia.
 Rapid increases in desflurane concentration lead to transient but sometimes worrisome elevations in heart rate, blood pressure, and catecholamine levels that are more pronounced than occur with isoflurane, particularly in patients with cardiovascular disease.
Rapid increases in desflurane concentration lead to transient but sometimes worrisome elevations in heart rate, blood pressure, and catecholamine levels that are more pronounced than occur with isoflurane, particularly in patients with cardiovascular disease.
 Nonpungency and rapid increases in alveolar anesthetic concentration make sevoflurane an excellent choice for smooth and rapid inhalation inductions in pediatric and adult patients.
Nonpungency and rapid increases in alveolar anesthetic concentration make sevoflurane an excellent choice for smooth and rapid inhalation inductions in pediatric and adult patients.
Inhalation Anesthetics: Introduction
Nitrous oxide, chloroform, and ether were the first universally accepted general anesthetics. Methoxyflurane and enflurane, two potent halogenated agents, were used for many years in North American anesthesia practice. Methoxyflurane was the most potent inhalation agent, but its high solubility and low vapor pressure yielded longer inductions and emergences. Up to 50% of it was metabolized by cytochrome P-450 (CYP) enzymes to free fluoride (F−), oxalic acid, and other nephrotoxic compounds. Prolonged anesthesia with methoxyflurane was associated with a vasopressin-resistant, high-output, renal failure that was most commonly seen when F− levels increased to greater than 50 μmol/L. Enflurane has a nonpungent odor and is nonflammable at clinical concentrations. It depresses myocardial contractility. It also increases the secretion of cerebrospinal fluid (CSF) and the resistance to CSF outflow. During deep anesthesia with hypocarbia electroencephalographic changes can progress to a spike-and-wave pattern producing tonic-clonic seizures. Because of these concerns, methoxyflurane and enflurane are no longer used.
Five inhalation agents continue to be used in clinical anesthesiology: nitrous oxide, halothane, isoflurane, desflurane, and sevoflurane.
The course of a general anesthetic can be divided into three phases: (1) induction, (2) maintenance, and (3) emergence. Inhalation anesthetics, such as halothane and sevoflurane, are particularly useful in the induction of pediatric patients in whom it may be difficult to start an intravenous line. Although adults are usually induced with intravenous agents, the nonpungency and rapid onset of sevoflurane make inhalation induction practical for them as well. Regardless of the patient’s age, anesthesia is often maintained with inhalation agents. Emergence depends primarily upon redistribution from the brain and pulmonary elimination of these agents.
Because of their unique route of administration, inhalation anesthetics have useful pharmacological properties not shared by other anesthetic agents. For instance, administration via the pulmonary circulation allows a more rapid appearance of the drug in arterial blood than intravenous administration.
Pharmacokinetics of Inhalation Anesthetics
Although the mechanism of action of inhalation anesthetics is complex, likely involving numerous membrane proteins and ion channels, it is clear that producing their ultimate effect depends on attainment of a therapeutic tissue concentration in the central nervous system (CNS). There are many steps in between the anesthetic vaporizer and the anesthetic’s deposition in the brain (Figure 8-1).
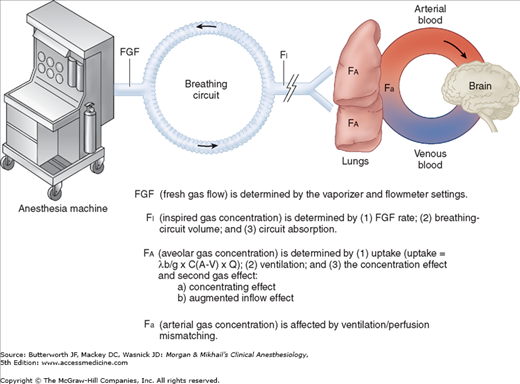
The fresh gas leaving the anesthesia machine mixes with gases in the breathing circuit before being inspired by the patient. Therefore, the patient is not necessarily receiving the concentration set on the vaporizer. The actual composition of the inspired gas mixture depends mainly on the fresh gas flow rate, the volume of the breathing system, and any absorption by the machine or breathing circuit. The higher the fresh gas flow rate, the smaller the breathing system volume, and the lower the circuit absorption, the closer the inspired gas concentration will be to the fresh gas concentration. Clinically, these attributes translate into faster induction and recovery times.
If there were no uptake of anesthetic agent by the body, the alveolar gas concentration (FA) would rapidly approach the inspired gas concentration (FI). Because anesthetic agents are taken up by the pulmonary circulation during induction, alveolar concentrations lag behind inspired concentrations (FA/FI <1.0). The greater the uptake, the slower the rate of rise of the alveolar concentration and the lower the FA:FI ratio.
Because the concentration of a gas is directly proportional to its partial pressure, the alveolar partial pressure will also be slow to rise. The alveolar partial pressure is important because it determines the partial pressure of anesthetic in the blood and, ultimately, in the brain. Similarly, the partial pressure of the anesthetic in the brain is directly proportional to its brain tissue concentration, which determines clinical effect.
 Therefore, the greater the uptake of anesthetic agent, the greater the difference between inspired and alveolar concentrations, and the slower the rate of induction.
Therefore, the greater the uptake of anesthetic agent, the greater the difference between inspired and alveolar concentrations, and the slower the rate of induction.
 Three factors affect anesthetic uptake: solubility in the blood, alveolar blood flow, and the difference in partial pressure between alveolar gas and venous blood.
Three factors affect anesthetic uptake: solubility in the blood, alveolar blood flow, and the difference in partial pressure between alveolar gas and venous blood.
Relatively soluble agents, such as nitrous oxide, are taken up by the blood less avidly than more soluble agents, such as halothane. As a consequence, the alveolar concentration of nitrous oxide rises faster than that of halothane, and induction is faster. The relative solubilities of an anesthetic in air, blood, and tissues are expressed as partition coefficients (Table 8-1). Each coefficient is the ratio of the concentrations of the anesthetic gas in each of two phases at steady state. Steady state is defined as equal partial pressures in the two phases. For instance, the blood/gas partition coefficient (λb/g) of nitrous oxide at 37°C is 0.47. In other words, at steady state, 1 mL of blood contains 0.47 as much nitrous oxide as does 1 mL of alveolar gas, even though the partial pressures are the same. Stated another way, blood has 47% of the capacity for nitrous oxide as alveolar gas. Nitrous oxide is much less soluble in blood than is halothane, which has a blood/gas partition coefficient at 37°C of 2.4. Thus, almost five times more halothane than nitrous oxide must be dissolved to raise the partial pressure of blood. The higher the blood/gas coefficient, the greater the anesthetic’s solubility and the greater its uptake by the pulmonary circulation. As a consequence of this increased solubility, alveolar partial pressure rises more slowly, and induction is prolonged. Because fat/blood partition coefficients are greater than 1, blood/gas solubility is increased by postprandial lipidemia and is decreased by anemia.
| Agent | Blood/Gas | Brain/Blood | Muscle/Blood | Fat/Blood |
|---|---|---|---|---|
| Nitrous oxide | 0.47 | 1.1 | 1.2 | 2.3 |
| Halothane | 2.4 | 2.9 | 3.5 | 60 |
| Isoflurane | 1.4 | 2.6 | 4.0 | 45 |
| Desflurane | 0.42 | 1.3 | 2.0 | 27 |
| Sevoflurane | 0.65 | 1.7 | 3.1 | 48 |
The second factor that affects uptake is alveolar blood flow, which—in the absence of pulmonary shunting—is essentially equal to cardiac output. If the cardiac output drops to zero, so will anesthetic uptake. As cardiac output increases, anesthetic uptake increases, the rise in alveolar partial pressure slows, and induction is delayed. The effect of changing cardiac output is less pronounced for insoluble anesthetics, as so little is taken up regardless of alveolar blood flow.  Low-output states predispose patients to overdosage with soluble agents, as the rate of rise in alveolar concentrations will be markedly increased.
Low-output states predispose patients to overdosage with soluble agents, as the rate of rise in alveolar concentrations will be markedly increased.
The final factor affecting uptake of anesthetic by the pulmonary circulation is the partial pressure difference between alveolar gas and venous blood. This gradient depends on tissue uptake. If anesthetics did not pass into organs such as the brain, venous and alveolar partial pressures would become identical, and there would be no pulmonary uptake. The transfer of anesthetic from blood to tissues is determined by three factors analogous to systemic uptake: tissue solubility of the agent (tissue/blood partition coefficient), tissue blood flow, and the difference in partial pressure between arterial blood and the tissue.
To better understand inhaled anesthetic uptake and distribution, tissues have been classified into four groups based on their solubility and blood flow (Table 8-2). The highly perfused vessel-rich group (brain, heart, liver, kidney, and endocrine organs) is the first to encounter appreciable amounts of anesthetic. Moderate solubility and small volume limit the capacity of this group, so it is also the first to reach steady state (ie, arterial and tissue partial pressures are equal). The muscle group (skin and muscle) is not as well perfused, so uptake is slower. In addition, it has a greater capacity due to a larger volume, and uptake will be sustained for hours. Perfusion of the fat group nearly equals that of the muscle group, but the tremendous solubility of anesthetic in fat leads to a total capacity (tissue/blood solubility × tissue volume) that would take days to approach steady state. The minimal perfusion of the vessel-poor group (bones, ligaments, teeth, hair, and cartilage) results in insignificant uptake.
| Characteristic | Vessel Rich | Muscle | Fat | Vessel Poor |
|---|---|---|---|---|
| Percentage of body weight | 10 | 50 | 20 | 20 |
| Percentage of cardiac output | 75 | 19 | 6 | 0 |
| Perfusion (mL/min/100 g) | 75 | 3 | 3 | 0 |
| Relative solubility | 1 | 1 | 20 | 0 |
Anesthetic uptake produces a characteristic curve that relates the rise in alveolar concentration to time (Figure 8-2). The shape of this graph is determined by the uptakes of individual tissue groups (Figure 8-3). The initial steep rate of uptake is due to unopposed filling of the alveoli by ventilation. The rate of rise slows as the vessel-rich group—and eventually the muscle group—approach steady state levels of saturation.
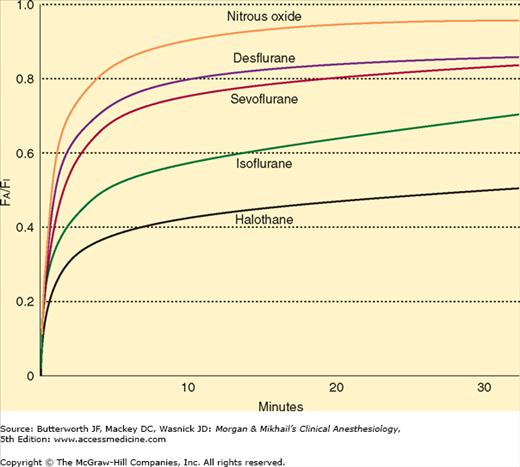
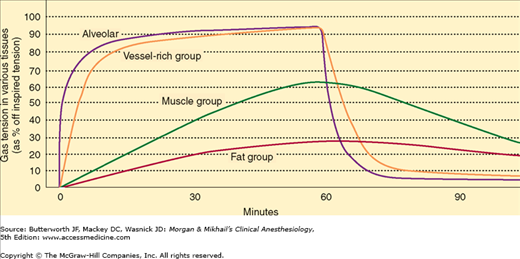
The lowering of alveolar partial pressure by uptake can be countered by increasing alveolar ventilation. In other words, constantly replacing anesthetic taken up by the pulmonary bloodstream results in better maintenance of alveolar concentration. The effect of increasing ventilation will be most obvious in raising the FA/FI for soluble anesthetics, as they are more subject to uptake. Because the FA/FI very rapidly approaches 1.0 for insoluble agents, increasing ventilation has minimal effect. In contrast to the effect of anesthetics on cardiac output, anesthetics that depress spontaneous ventilation (eg, ether or halothane) will decrease the rate of rise in alveolar concentration and create a negative feedback loop.
The slowing of induction due to uptake from alveolar gas can be reduced by increasing the inspired concentration. Interestingly, increasing the inspired concentration not only increases the alveolar concentration, but also increases its rate of rise (ie, increases FA/FI), because of two phenomena (see Figure 8-1) that produce a so-called “concentrating effect.” First, if 50% of an anesthetic is taken up by the pulmonary circulation, an inspired concentration of 20% (20 parts of anesthetic per 100 parts of gas) will result in an alveolar concentration of 11% (10 parts of anesthetic remaining in a total volume of 90 parts of gas). On the other hand, if the inspired concentration is raised to 80% (80 parts of anesthetic per 100 parts of gas), the alveolar concentration will be 67% (40 parts of anesthetic remaining in a total volume of 60 parts of gas). Thus, even though 50% of the anesthetic is taken up in both examples, a higher inspired concentration results in a disproportionately higher alveolar concentration. In this example, increasing the inspired concentration 4-fold results in a 6-fold increase in alveolar concentration. The extreme case is an inspired concentration of 100% (100 parts of 100), which, despite a 50% uptake, will result in an alveolar concentration of 100% (50 parts of anesthetic remaining in a total volume of 50 parts of gas).
The second phenomenon responsible for the concentration effect is the augmented inflow effect. Using the example above, the 10 parts of absorbed gas must be replaced by an equal volume of the 20% mixture to prevent alveolar collapse. Thus, the alveolar concentration becomes 12% (10 plus 2 parts of anesthetic in a total of 100 parts of gas). In contrast, after absorption of 50% of the anesthetic in the 80% gas mixture, 40 parts of 80% gas must be inspired. This further increases the alveolar concentration from 67% to 72% (40 plus 32 parts of anesthetic in a volume of 100 parts of gas).
The concentration effect is more significant with nitrous oxide, than with the volatile anesthetics, as the former can be used in much higher concentrations. Nonetheless, a high concentration of nitrous oxide will augment (by the same mechanism) not only its own uptake, but theoretically that of a concurrently administered volatile anesthetic. The concentration effect of one gas upon another is called the second gas effect, which is probably insignificant in the clinical practice of anesthesiology.
Normally, alveolar and arterial anesthetic partial pressures are assumed to be equal, but in fact, the arterial partial pressure is consistently less than end-expiratory gas would predict. Reasons for this may include venous admixture, alveolar dead space, and nonuniform alveolar gas distribution. Furthermore, the existence of ventilation/perfusion mismatching will increase the alveolar-arterial difference. Mismatch acts as a restriction to flow: It raises the pressure in front of the restriction, lowers the pressure beyond the restriction, and reduces the flow through the restriction. The overall effect is an increase in the alveolar partial pressure (particularly for highly soluble agents) and a decrease in the arterial partial pressure (particularly for poorly soluble agents). Thus, a bronchial intubation or a right-to-left intracardiac shunt will slow the rate of induction with nitrous oxide more than with halothane.
Recovery from anesthesia depends on lowering the concentration of anesthetic in brain tissue. Anesthetics can be eliminated by biotransformation, transcutaneous loss, or exhalation. Biotransformation usually accounts for a minimal increase in the rate of decline of alveolar partial pressure. Its greatest impact is on the elimination of soluble anesthetics that undergo extensive metabolism (eg, methoxyflurane). The greater biotransformation of halothane compared with isoflurane accounts for halothane’s faster elimination, even though it is more soluble. The CYP group of isozymes (specifically CYP 2EI) seems to be important in the metabolism of some volatile anesthetics. Diffusion of anesthetic through the skin is insignificant.
The most important route for elimination of inhalation anesthetics is the alveolus.  Many of the factors that speed induction also speed recovery: elimination of rebreathing, high fresh gas flows, low anesthetic-circuit volume, low absorption by the anesthetic circuit, decreased solubility, high cerebral blood flow (CBF), and increased ventilation. Elimination of nitrous oxide is so rapid that alveolar oxygen and CO2 are diluted. The resulting diffusion hypoxia is prevented by administering 100% oxygen for 5-10 min after discontinuing nitrous oxide. The rate of recovery is usually faster than induction because tissues that have not reached equilibrium will continue to take up anesthetic until the alveolar partial pressure falls below the tissue partial pressure. For instance, fat will continue to take up anesthetic and hasten recovery until the partial pressure exceeds the alveolar partial pressure. This redistribution is not as useful after prolonged anesthesia (fat partial pressures of anesthetic will have come “closer” to arterial partial pressures at the time the anesthetic was removed from fresh gas)—thus, the speed of recovery also depends on the length of time the anesthetic has been administered.
Many of the factors that speed induction also speed recovery: elimination of rebreathing, high fresh gas flows, low anesthetic-circuit volume, low absorption by the anesthetic circuit, decreased solubility, high cerebral blood flow (CBF), and increased ventilation. Elimination of nitrous oxide is so rapid that alveolar oxygen and CO2 are diluted. The resulting diffusion hypoxia is prevented by administering 100% oxygen for 5-10 min after discontinuing nitrous oxide. The rate of recovery is usually faster than induction because tissues that have not reached equilibrium will continue to take up anesthetic until the alveolar partial pressure falls below the tissue partial pressure. For instance, fat will continue to take up anesthetic and hasten recovery until the partial pressure exceeds the alveolar partial pressure. This redistribution is not as useful after prolonged anesthesia (fat partial pressures of anesthetic will have come “closer” to arterial partial pressures at the time the anesthetic was removed from fresh gas)—thus, the speed of recovery also depends on the length of time the anesthetic has been administered.
Pharmacodynamics of Inhalation Anesthetics
General anesthesia is an altered physiological state characterized by reversible loss of consciousness, analgesia, amnesia, and some degree of muscle relaxation. The multitude of substances capable of producing general anesthesia is remarkable: inert elements (xenon), simple inorganic compounds (nitrous oxide), halogenated hydrocarbons (halothane), ethers (isoflurane, sevoflurane, desflurane), and complex organic structures (propofol). A unifying theory explaining anesthetic action would have to accommodate this diversity of structure. In fact, the various agents probably produce anesthesia by differing sets of molecular mechanisms. Inhalational agents interact with numerous ion channels present in the CNS and peripheral nervous system. Nitrous oxide and xenon are believed to inhibit N-methyl-D-aspartate (NMDA) receptors. NMDA receptors are excitatory receptors in the brain. Other inhalational agents may interact at other receptors (eg, gamma-aminobutyric acid [GABA]-activated chloride channel conductance) leading to anesthetic effects. Additionally, some studies suggest that inhalational agents continue to act in a nonspecific manner, thereby affecting the membrane bilayer. It is possible that inhalational anesthetics act on multiple protein receptors that block excitatory channels and promote the activity of inhibitory channels affecting neuronal activity, as well as by some nonspecific membrane effects.
There does not seem to be a single macroscopic site of action that is shared by all inhalation agents. Specific brain areas affected by various anesthetics include the reticular activating system, the cerebral cortex, the cuneate nucleus, the olfactory cortex, and the hippocampus; however, to be clear, general anesthetics bind throughout the CNS. Anesthetics have also been shown to depress excitatory transmission in the spinal cord, particularly at the level of the dorsal horn interneurons that are involved in pain transmission. Differing aspects of anesthesia may be related to different sites of anesthetic action. For example, unconsciousness and amnesia are probably mediated by cortical anesthetic action, whereas the suppression of purposeful withdrawal from pain may be related to subcortical structures, such as the spinal cord or brain stem. One study in rats revealed that removal of the cerebral cortex did not alter the potency of the anesthetic! Indeed, measures of minimal alveolar concentration (MAC), the anesthetic concentration that prevents movement in 50% of subjects or animals, are dependent upon anesthetic effects at the spinal cord and not at the cortex.
 Past understanding of anesthetic action attempted to identify a unitary hypothesis of anesthetic effects. This hypothesis proposes that all inhalation agents share a common mechanism of action at the molecular level. This was previously supported by the observation that the anesthetic potency of inhalation agents correlates directly with their lipid solubility (Meyer-Overton rule). The implication is that anesthesia results from molecules dissolving at specific lipophilic sites. Of course, not all lipid-soluble molecules are anesthetics (some are actually convulsants), and the correlation between anesthetic potency and lipid solubility is only approximate (Figure 8-4).
Past understanding of anesthetic action attempted to identify a unitary hypothesis of anesthetic effects. This hypothesis proposes that all inhalation agents share a common mechanism of action at the molecular level. This was previously supported by the observation that the anesthetic potency of inhalation agents correlates directly with their lipid solubility (Meyer-Overton rule). The implication is that anesthesia results from molecules dissolving at specific lipophilic sites. Of course, not all lipid-soluble molecules are anesthetics (some are actually convulsants), and the correlation between anesthetic potency and lipid solubility is only approximate (Figure 8-4).
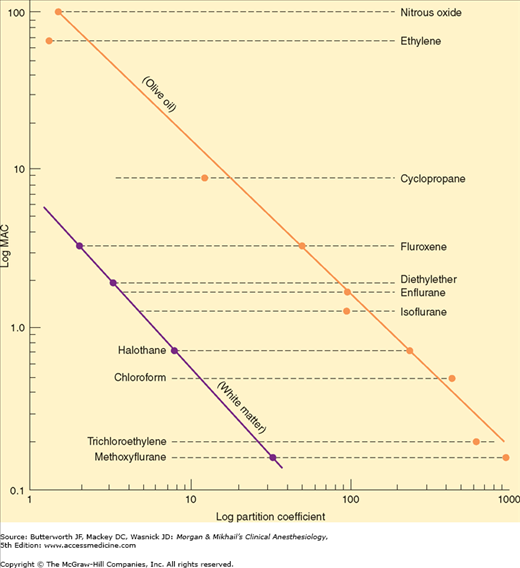
Neuronal membranes contain a multitude of hydrophobic sites in their phospholipid bilayer. Anesthetic binding to these sites could expand the bilayer beyond a critical amount, altering membrane function (critical volume hypothesis). Although this theory is almost certainly an oversimplification, it explains an interesting phenomenon: the reversal of anesthesia by increased pressure. Laboratory animals exposed to elevated hydrostatic pressure develop a resistance to anesthetic effects. Perhaps the pressure is displacing a number of molecules from the membrane or distorting the anesthetic binding sites in the membrane, increasing anesthetic requirements. However, studies in the 1980s demonstrated the ability of anesthetics to inhibit protein actions, shifting attention to the numerous ion channels that might affect neuronal transmission and away from the critical volume hypothesis.
General anesthetic action could be due to alterations in any one (or a combination) of several cellular systems, including voltage-gated ion channels, ligand-gated ion channels, second messenger functions, or neurotransmitter receptors. For example, many anesthetics enhance GABA inhibition of the CNS. Furthermore, GABA receptor agonists seem to enhance anesthesia, whereas GABA antagonists reverse some anesthetic effects. There seems to be a strong correlation between anesthetic potency and potentiation of GABA receptor activity. Thus, anesthetic action may relate to binding in relatively hydrophobic domains in channel proteins (GABA receptors). Modulation of GABA function may prove to be a principal mechanism of action for many anesthetic drugs.
The glycine receptor α1-subunit, whose function is enhanced by inhalation anesthetics, is another potential anesthetic site of action.
The tertiary and quaternary structure of amino acids within an anesthetic-binding pocket could be modified by inhalation agents, perturbing the receptor itself, or indirectly producing an effect at a distant site.
Other ligand-gated ion channels whose modulation may play a role in anesthetic action include nicotinic acetylcholine receptors and NMDA receptors.
Investigations into mechanisms of anesthetic action are likely to remain ongoing for many years, as many protein channels may be affected by individual anesthetic agents, and no obligatory site has yet been identified. Selecting among so many molecular targets for the one(s) that provide optimum effects with minimal adverse actions will be the challenge in designing better inhalational agents.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


