Increased Intracranial Pressure
Cheri A. Sulek
CASE SUMMARY
A 70-year-old, right-handed man presented to the emergency department with a history of confusion, intermittent slurred speech, difficulty with expressing and understanding speech, and progressive headaches. At the onset of his symptoms 3 months ago, he was evaluated at an outside hospital and diagnosed with a left-sided stroke and possible seizure disorder. The etiology of his stroke was not determined, and he was discharged home on anticonvulsants. He improved with physical therapy after discharge; however, his family noted a progressive decline in language function and worsening headaches. His medical history was significant for a myocardial infarction status post coronary artery bypass grafting, hypertension, smoking, and hyperlipidemia. Magnetic resonance imaging (MRI) of the brain with contrast revealed a multilobulated increasing mass in the left temporal and parietal lobes, measuring 5 × 5 cm, with a large amount of peritumor edema. A significant mass effect was noted on the left lateral ventricle and an 8-mm left-to-right shift, with early transtentorial herniation. He was started on dexamethasone, with significant improvement in his headaches, and remained on anticonvulsant therapy. He underwent a left craniotomy for biopsy and tumor debulking under general anesthesia without complications. Pathology was consistent with glioblastoma multiforme.
INTRODUCTION
Maintenance of normal intracranial pressure (ICP) is determined and closely regulated by well-defined structural components of the intracranial compartment. The introduction of intracranial pathology will ultimately exhaust the mechanisms that maintain cerebral homeostasis and lead to elevated ICP and abnormal intracranial elastance. The concept of abnormal ICP and its management goals are not static, and change as we gain a better appreciation of the underlying pathophysiologic processes involved, the impact of secondary neuronal injury, and the information obtained from cerebral monitoring available today. It is important to recognize the impact of hemodynamic manipulations, pharmacologic interventions, anesthetic agents, and ventilation on ICP and cerebral perfusion pressure (CPP). The goals of this chapter are to provide an understanding of : (i) ICP and its determinants; (ii) the regulation of ICP and its effects on CPP in normal and pathologic states; (iii) the impact of anesthetic agents on ICP; and (iv) the management of intracranial hypertension.
What Are the Structures that Comprise the Intracranial Compartment?
The intracranial compartment is defined by its contents: Brain tissue, cerebrospinal fluid (CSF), blood, and meninges. They are encased by the calvarium and communicate with the spinal axis through the foramen magnum. In the absence of pathology, the intracranial volume remains constant within the neuraxis. The calvarium is a bony, nondistensible, semiclosed container that strictly limits both intracranial volume and any expansion by acute or chronic pathologic processes.
Brain tissue alone accounts for 88% of the total intracranial volume. Eighty percent of the brain volume is water, and 20% of this is sequestered extracellularly.1 This extracellular environment is tightly regulated by an intact blood-brain barrier, which selectively permits diffusion and active transport of limited substances.
CSF accounts for 9% of the total intracranial volume.2 The total CSF volume is 150 mL in a normal adult, with daily production of 500 to 600 mL and replacement every 8 hours.3 Fifty percent of the total CSF volume (75 mL) is contained within the intracranial space.4 CSF is secreted primarily by the choroid plexus lining portions of the ventricular system and is principally absorbed from the subarachnoid space into the venous blood at the arachnoid granulations bordering the superior sagittal sinus. Obstructions of any portion of the ventricular system, including the arachnoid granulations, results in hydrocephalus and, occasionally, interstitial edema.
The cerebral vascular system is the smallest component of the intracranial space, contributing only 2% to 3% of its volume.2 The brain receives 15% of the cardiac output, with a rapid transit time. Cerebral blood volume (CBV) is only 75 mL, with three quarters residing within the low-pressure venous system.5 Despite its small volume, the vascular space is an important determinant of ICP and the parameter most affected by ventilation, anesthetic agents, and vasodilating drugs.
Intracranial volume remains constant until a new volume is added or expansion of an existing intracranial volume occurs in the nondistensible system. For ICP to remain normal, a mandatory reciprocal reduction in volume of one of the intracranial components (i.e., blood, CSF) must occur to offset the effects of the new volume added. These concepts constitute the principles of the Monroe-Kellie doctrine and its modifications.6
When Is Intracranial Pressure Considered Abnormal?
Normal ICP measured in adults in the supine position is 5 to 15 mmHg; normal values are 1.5 to 6 mmHg in infants and 3 to 7 mmHg in young children.7 In the supine position, pressures are considered equal within the craniospinal axis. Lumbar subarachnoid pressures reflect ICP as long as the two compartments freely communicate and no obstruction exists. It is now recognized that ICP is not always uniform within the craniospinal axis when pathology is present. Local differences in ICP and cerebral blood flow (CBF) often exist in areas of pathology (i.e., brain tumors or traumatic brain injury [TBI]), although, global ICP may be recorded as normal and is misleading.8,9 ICP is considered abnormal with levels of 15 mmHg in infants, 18 mmHg in children younger than 8 years, and 20 mm Hg in older children and adults.7 Neurologic outcome, especially in head-injured patients, appears to correlate with the degree and duration of intracranial hypertension.10 Sustained elevations in ICP that exceed 25 to 30 mmHg are frequently associated with poor outcome or are fatal. The detrimental effects of intracranial hypertension are the result of cerebral ischemia and the direct compressive effects on cerebral structures.
What Are the Mechanisms of Spatial Compensation?
ICP can be maintained at normal levels and regulated, even in the presence of a space-occupying lesion, as long as compensatory mechanisms are operational and the pathologic process evolves slowly. If rapid volume expansion occurs (i.e., epidural hematoma), ICP rises acutely because of inadequate compensatory mechanisms. A chronic pathologic process such as a slow-growing tumor will allow adequate reductions in cerebral blood and CSF volume to maintain normal intracranial dynamics until the mechanisms for spatial compensation are exhausted. The CSF system has the greatest buffering capacity of the intracranial contents, largely because of CSF absorption. Intracranial hypertension from a chronic pathologic process occurs when the buffering capacity of the CSF pathways is exhausted.
How Is Intracranial Elastance Defined?
The intracranial pressure-volume curve was defined by Langfitt et al. in 196511 (see Fig. 22.1). During the period of
compensation (horizontal axis), ICP increases minimally, even with relatively large changes in intracranial volume. When the buffering capacity of the intracranial space is exhausted, small increases in volume are followed by abrupt rises in ICP. If a mass expands rapidly, the additional volume cannot be accommodated, and ICP rises acutely.
compensation (horizontal axis), ICP increases minimally, even with relatively large changes in intracranial volume. When the buffering capacity of the intracranial space is exhausted, small increases in volume are followed by abrupt rises in ICP. If a mass expands rapidly, the additional volume cannot be accommodated, and ICP rises acutely.
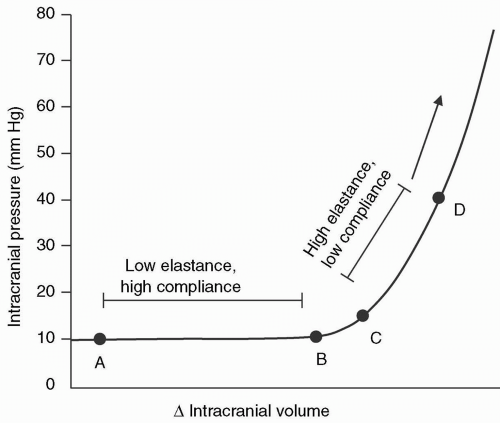 FIGURE 22.1 Intracranial pressure-volume curve. Along the horizontal axis (points A-B), a change in intracranial volume is offset by a reciprocal decrease in another intracranial component, allowing for spatial compensation. Large changes in intracranial volume can occur without a change in intracranial pressure. At point C, compensatory mechanisms are exhausted. Between points C-D, small changes in intracranial volume result in large changes in intracranial pressure. (Modified from: Fig. 44.1 in Kirby RR, Gravenstein N, Lobato EB, et al., eds. Clinical anesthesia practice, 2nd ed. WB Saunders; 2002:836.) |
The slope dP/dV of the pressure-volume curve defines the elastance of the system and reflects its stiffness or resistance to the deformation exerted by the intracranial contents with the addition of volume.12 Compliance is also used to define the pressure-volume curve; however, elastance more accurately defines this relation. Compliance is defined as the slope dV/dP and reflects the distensibility of the intracranial space.
How Is Cerebral Perfusion Pressure Defined?
The relation between ICP and CPP is defined by the following formula: CPP = MAP-ICP (MAP = mean arterial pressure). The lower limit of CPP considered acceptable in normotensive adults is 50 mmHg. Under normal conditions, ICP is usually 5 mmHg or less, and CPP changes reflect alterations in MAP. Central venous pressure has been used when ICP is unknown or not measured. In the presence of intracranial pathology, effective perfusion pressure declines as ICP rises. CPP should be maintained at >60 mmHg in head-injured patients, and perhaps higher when there is documented global or focal ischemia.13 A decrease in MAP or an elevation in ICP will deleteriously alter the effective perfusion pressure.
How Is Cerebral Blood Flow Regulated?
Cerebral autoregulation involves the maintenance of a constant CBF when MAP (or CPP if ICP is normal) is between 50 and 150 mm Hg and is accomplished by adjusting the cerebrovascular resistance (CVR) (see Fig. 22.2). In recent years, the lower limit of autoregulation has been questioned and is likely higher than previously depicted by Lassen.14 There is evidence that the lower limit of autoregulation in nonanesthetized, normotensive adults is as high as 70 mmHg and that the lower limit depicted in the autoregulation curve may be more applicable to anesthetized patients or those receiving vasodilating agents that lower the limit of autoregulation (i.e., sodium nitroprusside).15 When the limits of autoregulation are impaired or exceeded, CBF passively follows blood pressure. In chronic hypertension, the autoregulatory curve is shifted to the right; consequently, in hypertensive individuals, cerebral ischemia can occur at a CPP that generally would ensure adequate CBF in normotensive subjects. Intracranial hypertension alone does not appear to abolish autoregulation. Autoregulation of CBF occurs within seconds, and, at least in nonanesthetized humans, depends on the underlying PACO2.16 The exact mechanism of autoregulation is not known, but it is speculated that the resistance vessels may be responsive to the local metabolic environment or to transmural pressure.17
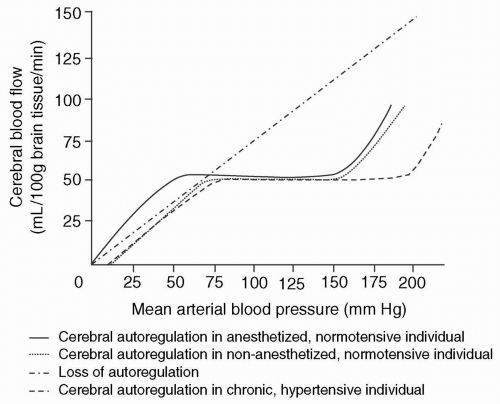 FIGURE 22.2 Cerebral autoregulation curve. Cerebral autoregulation is the maintenance of a constant cerebral blood flow when mean arterial pressure is 50 to 150 mmHg by adjustment of CVR. The lower limit of autoregulation has been questioned, and the classic lower limit of 50 mm Hg may reflect the anesthetized, normotensive patient. In poorly controlled hypertension, the autoregulation curve is shifted to the right so that cerebral ischemia may occur at a normal mean arterial pressure. (Modified from Fig. 44.2 in Kirby RR, Gravenstein N, Lobato EB, et al., eds. Clinical anesthesia practice, 2nd ed. WB Saunders; 2002:836.) |
Normal global CBF is approximately 50 to 55 mL/100 gm brain tissue/minute.18 Grey matter receives 75% of CBF, reflecting its increased metabolic activity compared to the white matter.19 Local increases in CBF in the nonpathologic state indicate the use of a specific region of the cortex during a task or activity.20 CBF is normally tightly coupled to cerebral oxygen consumption (CMRO2) and is expressed as CMRO2 = CBF × AVDO2 (arteriovenous difference of oxygen).21 Normally CMRO2 is constant at 3.2 to 3.5 mL/100gm brain tissue/minute.21 CBF and CMRO2 do not always remain coupled, as often observed after head injury and during the use of potent inhalational agents.22
Cerebral ischemia occurs when CBF falls below the critical level required to meet the metabolic demands of the cerebral tissue. The threshold for cerebral ischemia is increased under most general anesthetic techniques, especially isoflurane, and predominantly results from reduction in CMRO2.
What Factors Influence Cerebral Blood Flow and Cerebral Blood Volume?
CBF is influenced by pathology disrupting autoregulation, potent inhalational agents, intravenous anesthetics, and alterations in PACO2 and PaO2. In general, changes in CBF are accompanied by parallel changes in CBV at a ratio of 7:1.23 This relation does not always exist, and each factor affecting CBF and CBV must be considered independently. For example, inhalational agents and hypercarbia produce elevations in CBF and CBV by cerebral vasodilation, but hypotension with a MAP <50 mmHg (or higher in nonanesthetized individuals) results in decreased CBF but increased CBV, reflecting changes in the tone of the cerebral resistance vessels.
The cerebral resistance vessels, predominantly end-arterioles, are exquisitely sensitive to alterations in PACO2 and rapidly alter ICP. Hypocapnea is characterized by vasoconstriction of arterioles, resulting in decreased CBV, CBF, and ICP, whereas hypercapnea influences the vascular tone in opposite manner. CBF is increased by approximately 2 mL/100 gm brain tissue/minute or a 4% change for each mm Hg increase in PACO2.24 The pH of the extracellular fluid of the brain influences the tone of the cerebral vasculature, but within 24 to 36 hours, the CSF pH begins to normalize, and benefits of hyperventilation are lost.25 The responsiveness of the resistance vessels to carbondioxide (CO2) diminishes as the upper and lower limits of cerebral autoregulation are approached.24 A linear relation between CBF and PACO2 exists when PACO2 ranges from 20 to 80 mmHg.24 The potent inhalational anesthetic agents do not interfere with the reactivity of the cerebral vasculature to CO2 at a 1.0 minimum alveolar concentration (MAC) of anesthesia. CBF is not affected until the PaO2 falls below 50 mmHg, and reflects a response to tissue acidosis, but rapidly reverses when PaO2 is corrected.
How Is Intracranial Hypertension Clinically Diagnosed?
The symptoms and signs of elevated ICP are relatively nonspecific and depend largely on the chronicity and location of the underlying pathologic process. The clinical presentation of a patient with a chronic, slow-developing, intracranial process is vastly different from an acute process with early spatial exhaustion and impending herniation of brain tissue. The classic clinical manifestations of elevated ICP are headache, nausea, vomiting, visual disturbances, altered mentation, papilledema, and ocular palsies. Focal neurologic deficits reflect mass effect or ischemia of surrounding brain tissue by an expanding lesion. Headache frequently occurs and is located bifrontal or bioccipital; it is worse in the morning upon awakening, exacerbated by coughing or Valsalva maneuver, paroxysmal in nature, and relieved by emesis. Dural stretching and traction on vessels at the base of the brain caused by the mass lesion contribute to headache. Headache suggests elevated ICP but is not diagnostic. Nausea and projectile vomiting occur usually in the morning or at night, and emesis often relieves headache. Papilledema is present bilaterally 24 to 48 hours after ICP becomes elevated, and results from axoplasmic stasis as ICP is transmitted through the subarachnoid space to the optic disc. Focal neurologic deficits may be evident if an intracranial mass lesion compresses or distorts underlying sensory, motor, or cranial nerve pathways.
Deterioration in the neurologic examination progresses in a rostral-to-caudal fashion with supratentorial mass lesions. Brainstem dysfunction is a late finding with a slowly expanding mass, but occurs early with a rapidly enlarging mass and signals herniation.26 Conversely, the initial clinical manifestation of an infratentorial mass lesion may be brainstem dysfunction (i.e., cranial nerve palsy, cerebellar findings, respiratory abnormalities). Loss of consciousness associated with a supratentorial mass is ominous in the absence of a metabolic etiology and implies bilateral cerebral hemispheric or diencephalic dysfunction.
The degree of midline shift of the pineal body measured by computed tomography (CT) of the brain correlates with level of consciousness: Alert with a 0 to 3mm shift; drowsy with a 3 to 4 mm shift; stuporous with a 6 to 8.5 mm shift; and comatose with a 8 to 13 mm shift.27 Mental status impairment with infratentorial lesions often results from the development of noncommunicating hydrocephalus, but may be ominous and signal disruption or compression of the reticular activating system.
Assessment of brainstem function is the basis of the neurologic examination in a comatose patient. Evaluation of brainstem reflexes allows documentation of progressive neurologic deterioration and confirms brain death. The neurologic examination of brainstem function consists of assessment of pupil reactivity and size, respiratory pattern, oculomotor response, and motor response to painful stimuli.
What Neuroradiologic Imaging Studies Are Needed in a Patient with Intracranial Hypertension?
Any patient with suspected intracranial pathology and increased ICP requires evaluation with CT or MRI of the brain. Peritumor edema, diffuse cerebral edema, subarachnoid blood, hydrocephalus, compressed ambient cisterns, midline shift, and the presence of mass lesions suggest elevated ICP but do not provide quantitative
information (see Fig. 22.3). In patients with severe head injury, the presence of midline shift and compressed ambient cisterns usually predict increased ICP or herniation.28 It is important to correlate neuroradiologic findings with the neurologic examination because the clinical presentation often reflects the chronicity of the underlying disease process.
information (see Fig. 22.3). In patients with severe head injury, the presence of midline shift and compressed ambient cisterns usually predict increased ICP or herniation.28 It is important to correlate neuroradiologic findings with the neurologic examination because the clinical presentation often reflects the chronicity of the underlying disease process.
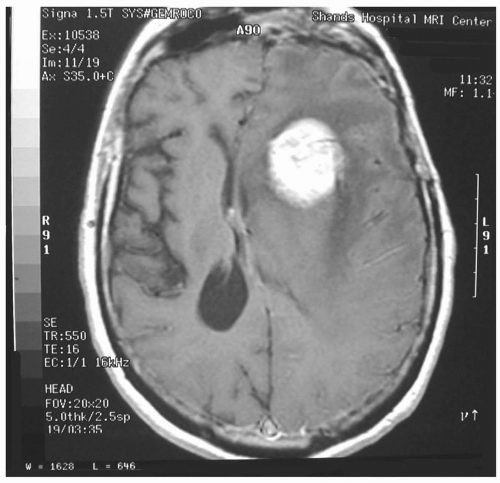 FIGURE 22.3 Magnetic resonance imaging of the brain depicting a large brain tumor with significant mass effect and edema formation. |
When Is Intracranial Pressure Monitoring Indicated and Which Device?
The intraventricular catheter (or ventriculostomy) was introduced in 1960 by Lundberg and has remained the gold standard for ICP monitoring.29 Newer monitoring modalities have been developed; ICP can be recorded from an epidural, subdural, subarachnoid, ventricular, or intraparenchymal location, depending upon the device used. The monitoring devices presently in use include catheters, bolts, screws, and fiberoptic cables. ICP monitoring is most often achieved from a supratentorial location and is less commonly employed in the posterior fossa due to risk of cranial nerve or brainstem injury.30 It is presumed that once the monitor is in position, the ICP recorded is reflective of pressure in the supratentorial and infratentorial compartments in the absence of obstruction; however, differences in ICP have been recorded not only between supratentorial and infratentorial compartments, but also within the same compartment (i.e., supratentorial).
▪ INTRACRANIAL PRESSURE MONITORS
An ICP monitor does not replace the neurologic examination but can guide treatment when the patient is comatose and intracranial hypertension is suspected. The Glasgow Coma Scale (GCS) is the gold standard utilized to determine the need for ICP monitoring in head-injured patients. A GCS of 8 or less in head-injured patients warrants the placement of a monitoring device.13 Any therapeutic intervention that may elevate ICP in a patient with compromised intracranial elastance warrants consideration of placement of an ICP monitor. The risks and benefits of ICP monitoring must be carefully weighed in the presence of coagulopathy. This issue has become even more relevant with the increased number of liver transplantations being performed, some of which will be done in patients with encephalopathy and coexisting coagulopathy. Indications for ICP monitoring and the device utilized often reflect the bias and experience of the neurosurgeon and any institutional protocols that exist.
There are several devices for clinical monitoring of ICP. The intraventricular catheter is considered the gold standard of recording devices and is used to determine the accuracy of new monitors. It has the dual advantage of ICP measurement and therapeutic CSF drainage to lower ICP. Technical problems limit its use in patients with small or compressed ventricles such as pseudotumor cerebri or a large brain tumor. Its placement requires more skill than with other monitoring devices and has been associated with a higher incidence of infection, with a greater potential to cause brain injury during placement.
The subarachnoid bolt or screw is associated with a lower incidence of infection, less potential for brain injury, and requires less skill to place. Mollman et al. determined that the subarachnoid bolt was less accurate for ICP monitoring when compared to either a subarachnoid catheter or ventriculostomy technique.31 When ICP is high, subarachnoid bolts are less reliable because of obstruction of the lumen of the bolt by brain tissue and its propensity to produce falsely low readings.
Fiberoptic ICP monitoring systems are used frequently and can be placed in the intraparenchymal, subdural, or intraventricular compartment. Fiberoptic monitors have proven to be reliable, accurate devices when compared with intraventricular monitors.32,33 The devices have major drawbacks including significant drift, inability to recalibrate in vivo, expense and fragility of the fiberoptic catheter. It is recommended that these devices be replaced every 5 days to minimize false readings and infectious complications.
What Is the Role of Transcranial Doppler?
Transcranial doppler (TCD) imaging provides an inexpensive, noninvasive bedside method to document severe
intracranial hypertension, brain death, and presence of cerebral vasospasm. TCD imaging is an ultrasound technique that determines blood flow velocity, direction of flow through a major cerebral artery, and reflects the status of the vascular resistance of the vessel. As ICP approaches diastolic arterial pressure levels, the diastolic flow velocity diminishes, approaches 0, and then reverses flow.34 Brain death has been associated with one of three TCD patterns: (i) diastolic flow reversal; (ii) small systolic spikes; or (iii) absence of signal.34 An absent signal should be interpreted cautiously and verified in other vessels. Information derived from TCD monitoring must be used in conjunction with the clinical examination (brainstem reflexes) when documenting brain death.
intracranial hypertension, brain death, and presence of cerebral vasospasm. TCD imaging is an ultrasound technique that determines blood flow velocity, direction of flow through a major cerebral artery, and reflects the status of the vascular resistance of the vessel. As ICP approaches diastolic arterial pressure levels, the diastolic flow velocity diminishes, approaches 0, and then reverses flow.34 Brain death has been associated with one of three TCD patterns: (i) diastolic flow reversal; (ii) small systolic spikes; or (iii) absence of signal.34 An absent signal should be interpreted cautiously and verified in other vessels. Information derived from TCD monitoring must be used in conjunction with the clinical examination (brainstem reflexes) when documenting brain death.
What Is the Role of Somatosensory and Brainstem Auditory-Evoked Potentials?
Multimodality-evoked potentials, including somatosensory and brainstem auditory-evoked potentials, can be used to diagnose brain death and predict neurologic outcome in comatose patients. Both types of evoked potential recording modalities are not accepted as the sole diagnostic criteria and should be used with knowledge of the neurologic examination. The absence of all waveforms, except wave I in brainstem auditory-evoked potential recording, accompanied by absence of cortical somatosensory-evoked potentials reliably predicts brain death.35 Wave I may be absent because of destruction of the organ of Corti or due to ototoxic drug administration. In the presence of normal or abnormal brainstem auditory-evoked potentials and absence of cortical somatosensory-evoked potentials, a persistent vegetative state is predicted.35
What Are the Effects of Anesthetics and Drugs on Intracranial Pressure?
The selection of an anesthetic technique and supplemental drugs for a patient with intracranial hypertension should be tailored to perform the following functions: (i) Maintain adequate CBF and CPP, (ii) minimize ICP elevations, (iii) provide cerebral protective measures, and (iv) reduce ICP. It is important for the anesthesiologist to recognize the impact that anesthetic agents or pharmacologic interventions may have in improving or worsening the primary underlying pathology. The use of agents to prevent or attenuate secondary neuronal injury following cerebral ischemia may become as important as the treatment directed toward the primary injury.
▪ INHALATION AGENTS
Halothane has limited use in modern anesthesia practice today and is used predominantly for children. In animals and humans, halothane is a potent cerebral vasodilator that increases CBF by reducing CVR and deleteriously affects ICP, particularly in the presence of an intracranial mass.36 It is well accepted that halothane increases CBF in a dose-related fashion, and most agree that it increases the CBF-to-CMRO2 ratio to a greater extent than other volatile agents in clinical use.37 The reactivity of the cerebral circulation to CO2 is preserved with halothane at 1.0 MAC anesthesia. Cerebral autoregulation is impaired to a greater extent with halothane than with isoflurane or enflurane.37 Autoregulation may be impaired but not abolished with halothane concentrations as low as 0.5%.38 The duration of impairment during anesthesia is not known. In current clinical practice, there are safer, alternative inhalational agents that can be substituted for halothane in patients with space-occupying lesions.
Isoflurane remains one of the safest, time-tested, volatile anesthetics used in patients with intracranial pathology. Isoflurane has profound nonlinear(at < 1.0 MAC anesthesia), dose-related, cerebral metabolic depressant properties. Increasing anesthetic depth is not associated with disruption of the cerebral metabolic pathways.39
Isoflurane is a moderate cerebral vasodilator that increases CBF in a dose-related manner in dogs, and is attributed to a decline in CVR.40 In humans, minimal CBF changes occur with isoflurane concentrations of 1% to 2%.41 Profound changes have been observed, however, when nitrous oxide is substituted for an equipotent concentration of isoflurane.41 Isoflurane may elevate ICP, especially in patients with increased intracranial elastance. Establishing hyperventilation (PACO2 25 to 30 mmHg) simultaneously with introduction of isoflurane consistently prevented elevations in CSF pressure in patients with intracranial pathology.42 Increases in ICP in response to isoflurane result from cerebral vasodilation alone. Resistance to reabsorption of CSF is lowered, and rate of CSF production is unaffected with isoflurane.43 Cerebral autoregulation remains intact at 1.0 MAC but is significantly attenuated or abolished at 2.0 MAC of isoflurane.44
Although isoflurane remains a safe volatile agent for use in intracranial procedures, the combination of nitrous oxide and isoflurane should be avoided in patients with existing intracranial disease, because elevations in CBF cannot be consistently prevented with hyperventilation.
Sevoflurane causes minimal effects on CBF even with high doses in animals.45,46 Sevoflurane has similar effects as isoflurane on ICP and CPP in humans with intracranial pathology.47 Sevoflurane may have epileptogenic activity similar to enflurane, a property that would make this agent undesirable in neurosurgical patients. From limited studies, sevoflurane may possess epileptogenic properties that are weaker and elicited by higher concentrations than enflurane. Sevoflurane should be safe in neuroanesthesia as long as low concentrations of volatile agent are used.
Sevoflurane closely resembles isoflurane in its effects on CBF, CMRO2 and CBV. The addition of nitrous oxide
to sevoflurane precipitously increases CBF and impairs autoregulation.48 The data is conflicting: Sevoflurane may precipitate seizure activity at high anesthetic concentrations but should be a safe volatile agent when used in clinically relevant concentrations.
to sevoflurane precipitously increases CBF and impairs autoregulation.48 The data is conflicting: Sevoflurane may precipitate seizure activity at high anesthetic concentrations but should be a safe volatile agent when used in clinically relevant concentrations.
Desflurane is a volatile anesthetic with a low blood-gas partition coefficient that allows for rapid emergence to evaluate the neurologic status of the patient. The effects of desflurane on CBF and ICP are conflicting, but there is evidence that it has significant cerebral vasodilating properties.49,50 CBF increases in a dose-dependent manner and is augmented by hemodynamic support in animals.50,51 Reductions in CMRO2 following electroencephalogram (EEG) changes are similar to isoflurane anesthetic.50 The response of the cerebral vasculature to CO2 is intact in dogs and humans at clinically relevant concentrations of desflurane.52
The effect of desflurane on ICP is controversial, particularly in humans. ICP elevation has been documented in canine and porcine studies, and are similar to those reported under isoflurane anesthesia in the absence of pathology.50,52 It is speculated that ICP elevations are attributable to an elevated CBV reflecting desflurane’s potent vasodilating properties. Because animal and human studies are conflicting, desflurane should probably be used cautiously in patients with abnormal intracranial elastance.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


