CHAPTER 39 Diuretics and Newer Therapies for Sodium and Edema Management in Acute Decompensated Heart Failure
FREQUENTLY, ACUTE cardiac decompensation is the clinical expression of left ventricular dysfunction, occurring as the result of diverse etiologies. Even at advanced stages, many patients initially thought to require transplantation due to acute decompensation, can continue to be managed with medical therapy. Many factors contribute to progression of heart failure, from mild forms to the advanced or refractory condition. However, the precise characteristics which govern the rate of progression will vary from patient to patient. As a result, the onset of decompensation or refractory presentation is difficult to predict, and may be abrupt in onset. In this regard, evidence of deterioration must be monitored carefully. For patients with chronic heart failure, fluid retention is typically considered the primary cause of abrupt decompensation, and surveys of hospital admissions support this observation. However, it is important to remember that fluid retention is just one of many factors that can result in abrupt decompensation (Table 39-1). The hallmark of acute cardiac decompensation is pulmonary edema, which results from excessive sodium retention, acute left ventricular dysfunction, or mechanical disorders of the left ventricle. Sodium retention results from the inability of the body to excrete sodium at a rate commensurate with dietary sodium intake. In fact, the characteristics which are identified as the hallmark of acute cardiac decompensation are for the most part findings of excess sodium retention: pulmonary congestion, peripheral edema, increased jugular venous pressure, an S3 gallop, and hepatomegaly/ascites. In addition to urgent care measures, the most common therapy of acute cardiac decompensation is the use of diuretics. In recent years, newer approaches to sodium and fluid management have been developed, which may improve clinical outcomes and reduce hospital morbidity during the time of acute decompensation. In this chapter, emphasis will be placed upon acute therapy with diuretics, and the transition to a more stable chronic regimen. Newer classes of therapy will also be highlighted.
Table 39–1 Conditions That May Be Characterized as Acute Decompensated Heart Failure
| Pulmonary edema |
| Progressive or persistent severe fluid retention |
| Fatigue or weakness with minimal exertion |
| Frequent nocturnal decompensation/dyspnea |
| LV dysfunction due to new or recurrent angina |
| Progressive renal failure of varying origins |
| Uncontrolled hypertension |
Decompensated Heart Failure and Edema
Mechanisms
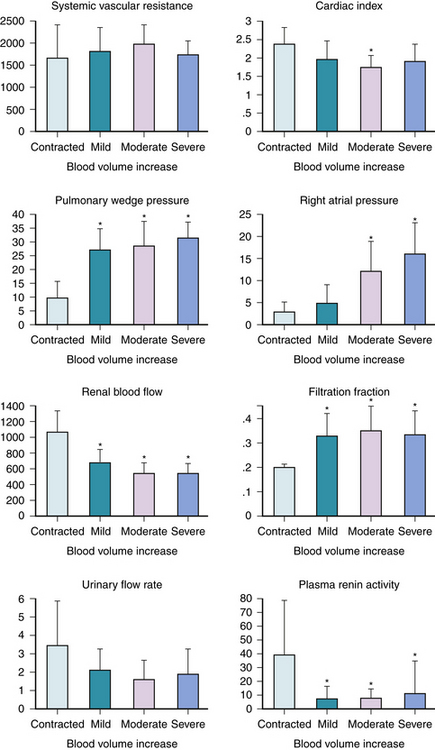
Cardiac decompensation is characterized by decreased left ventricular systolic function, and in most cases, abnormal diastolic function. The resultant decrease of cardiac output and increased ventricular end-diastolic pressure set the stage for sodium retention (Fig. 39-1). Decreased cardiac output and increased systemic resistance produce decreased renal blood flow, and magnitude of renal blood flow reduction is correlated with decreased cardiac output,1,2 and diminished renal blood flow is a stimulus for activation of neurohormonal vasoconstriction pathways, particularly the renin system,3,4 which produces vasoconstriction and aldosterone secretion. Total body water, extracellular fluid, and plasma volume are increased compared with controls.5 Even within the heart failure population, there may be a spectrum of blood volume expansion (Fig. 39-2). When this is moderate to severe, impaired renal function and increased cardiac filling pressures occur. These hemodynamic responses become the basis for sodium retention.
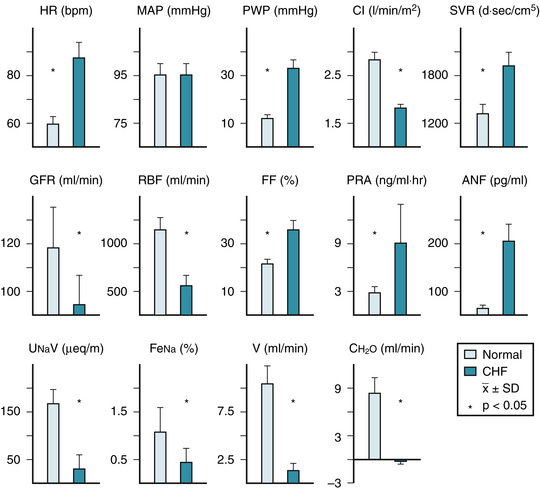
The reduction of the glomerular filtration rate in heart failure is highly correlated with hemodynamic parameters, where the greatest reduction of cardiac output and renal blood flow are associated with the greatest reduction of the glomerular filtration rate.1,6 Renal blood flow and function tend to decrease with age in normal subjects, and an age effect can be superimposed on the overall reduction in renal blood flow and function due to the heart failure process.6 The resultant diminished delivery of sodium to the distal nephron at the level of the macula densa becomes a potent stimulus for renin release.7 Aldosterone further increases sodium retention at the distal nephron at the expense of potassium excretion. Although current therapy for heart failure may improve cardiac output and renal blood flow, no current oral therapy has uniformly improved the glomerular filtration rate.
Neurohormonal factors that promote retention of sodium and water include aldosterone, vasopressin, angiotensin II, norepinephrine, and the vasoconstrictor prostaglandins; in contrast, prostacyclin, dopamine, and atrial natriuretic factor favor sodium excretion.3,4 The effect of vasopressin is primarily the promotion of free water retention, rather than sodium retention, and this typically contributes to hyponatremia.
Pharmacokinetics of Diuretic Therapy
Response to Diuretic Administration for Acute Cardiac Decompensation
The sites of diuretic actions and their role in the edema of cardiac decompensation have been reviewed.10,11 In the last decade, there have not been substantial breakthroughs in new diuretic development or mechanisms. The hemodynamic response to diuretic therapy has been characterized primarily in the setting of pulmonary edema. The acute hemodynamic response revealed a vasodilator effect12–14 and reduction of cardiac filling pressures.12–15 During acute decompensated heart failure, changes in cardiac output varied from increased or no change to significant reduction, and changes in hemodynamics could proceed, or lag, production of diuresis. Such evaluations were conducted in diverse patient populations and must be interpreted accordingly. It has been stated that the vasodilator effect could precede the occurrence of diuresis, but the database for this effect is limited. Studies have reported either vasoconstriction,16 or initial vasodilation followed by vasoconstriction17 with acute or short-term follow-up. A review of the cited literature therefore reveals a fully divergent range of hemodynamic response to diuretics, although the majority of acute studies demonstrate a reduction of cardiac filling pressures.12–15 Loop diuretics have been shown to decrease renal vasculature resistance and to increase total and cortical renal blood flow.13,18,19 However, this may be related to severity of heart failure.20 It is difficult to isolate a single neurohormonal factor as being primarily responsible for the overall hemodynamic and regional hemodynamic response to diuretics. Acute diuretic therapy may increase renin and aldosterone.16,17,20,21 Sympathetic nervous system activity also increases in response to diuretics.20,21 The impact of neurohormonal activation is an attenuation of the otherwise favorable effects of diuretics.
Loop Diuretics
As this chapter focuses on acute cardiac decompensation, the response to parenteral diuretic therapy will be emphasized (Table 39-2), without an extensive review of pharmacology. The term loop diuretic has evolved to encompass pharmacologic compounds which exert their primary action on the thick ascending loop of Henle. To reach the intraluminal site of action, these organic acids must first be secreted into the proximal tubule via the organic acid pathway. Once in the lumen, active reabsorption of chloride is inhibited in both the medullary and cortical portions of the loop of Henle. Decreased sodium reabsorption also occurs since the chloride ion is cotransported with sodium and potassium.
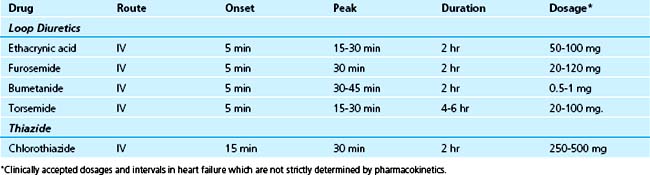
The three loop diuretics traditionally used in the United States to treat the edema of heart failure are furosemide, bumetanide, and torsemide. They are of equal efficacy but vary in pharmacokinetic properties.11,22 Ethacrynic acid remains available, but has a progressively diminishing role for edema management.23–25 Despite equal efficacy among loop diuretics, clinicians will often substitute one for the other with the hope of improved efficacy. Torsemide has a greater duration of action than its counterparts, but in general, the strategy of changing from one loop diuretic to another is typically not effective. After oral administration, furosemide has 40% bioavailability, bumetanide has 80%, and torsemide has greater than 80% bioavailability. A diminished diuresis and a prolonged renal elimination of the drug may be expected in heart failure when renal dysfunction is evident. Torsemide has two active metabolites, which probably accounts for its longer elimination half-life of 3 hours. Residual loop diuretic concentration is eliminated by nonrenal mechanisms, including hepatic degradation and excretion. Because of the differences in bioavailability and potency, equivalent doses of bumetanide, furosemide, and torsemide are 1 mg, 40 mg, and 20 mg, respectively. All three agents are extensively bound to plasma proteins and are rapidly secreted by the organic acid pathway of the proximal tubule. In chronic renal failure, competition for this pathway by exogenous and accumulated endogenous organic acids causes lower peak concentration of the drug at its site of action. Therefore, a diminished diuresis and a prolonged renal elimination of the drug may be expected in heart failure when renal dysfunction is evident. The diuretic effect is apparent within 30 minutes after oral administration and peaks in 1 to 2 hours.26–30 Bumetanide has a somewhat shorter duration of action than that of furosemide or torsemide. Torsemide, like other loop diuretics, must be secreted into the urine by the organic acid secretory system in the proximal tubule to be effective.23 It produces a maximum sodium excretion rate at approximately the same time with either intravenous or oral administration. Although the pharmacokinetics of torsemide are linear, increases in doses above 50 mg do not appear to increase the maximum sodium excretion rate but do increase the duration of the pharmacodynamic effect. One study has defined a ceiling dose of 100 mg of torsemide in patients with renal insufficiency and has recommended that repeated administration of torsemide is more effective than increasing the dose at this point.23 It has been shown in another study that 20 mg of intravenous torsemide is approximately as effective as 40 mg of intravenous furosemide.24 However, additional well-controlled trials of torsemide are necessary to determine whether it provides clinical benefit beyond current loop diuretics.
Thiazides and Potassium-Sparing Diuretics
Thiazide diuretics are derivatives of the benzothiadiazine structure.11,22 Like loop diuretics, they are organic acids and are highly protein-bound. Since they cannot be filtered, they gain access to the tubular lumen via the organic acid secretory pathway of the proximal tubule. Chlorothiazide is the only thiazide available in a soluble form that permits intravenous administration. This formulation has recently been discontinued and limited stores remain. Its appreciable benefits, combined with an intravenous loop diuretic, are now of historical interest. Oral hydrochlorothiazide or metolazone are effective in most cases, but delayed gastric and small bowel absorption are common in the presence of severe edema. Despite this limitation, the combination of loop and thiazide diuresis appreciably augments diuresis. Experimental animal data demonstrate reactive hypertrophy of the distal nephron during long-term sodium depletion.31
Related posts:
 Evolution of the Coronary Care Unit: Past, Present, and Future
Evolution of the Coronary Care Unit: Past, Present, and Future
 Elevated Cardiac Troponin in the Absence of Acute Coronary Syndromes: Mechanism, Significance, and Prognosis
Elevated Cardiac Troponin in the Absence of Acute Coronary Syndromes: Mechanism, Significance, and Prognosis
 Conduction Disturbances in Acute Myocardial Infarction
Conduction Disturbances in Acute Myocardial Infarction
 Pharmacologic Interactions in the CICU
Pharmacologic Interactions in the CICU
 Acute Aortic Syndromes: Diagnosis and Management
Acute Aortic Syndromes: Diagnosis and Management
 Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


