Demyelinating Disease/Multiple Sclerosis
Ann Higgins MS, RN, ANP
Multiple sclerosis (MS) is a chronic disease of the central nervous system (CNS) that typically appears in late adolescence or early adulthood. Although the etiology is unknown, it is believed to be autoimmune in nature, after a viral infection, with genetic and environmental factors involved. During what could be their most productive and active years, affected persons become partially or completely disabled; this has a profound social, emotional, and financial impact on their lives.
The neurologic abnormalities that constitute MS may be acute or subacute in onset and may wax and wane over time. Early symptoms include numbness, double vision, paresis, bladder control problems, ataxia, and tremor. The clinical course of the disease may be benign, relapsing and remitting, or progressive. Treatment is based on the progression of the disease and symptoms, including fatigue, spasticity, urinary dysfunction, emotional problems, cognitive dysfunction, and pain. Management of chronic symptoms helps the patient remain functional even when the problems are severe. Medication designed to reduce a specific immune response and medications that assist in the stimulation of remyelination are being developed.
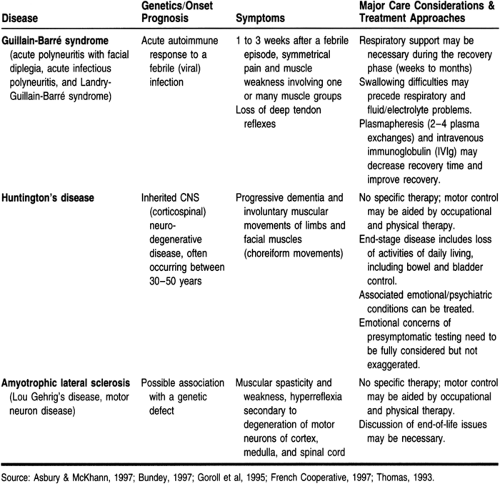
Other neurologic conditions, not emphasized in this chapter, may be similar in some ways to MS. Table 52-1 compares Guillain-Barré syndrome, Huntington’s disease, and amyotrophic lateral sclerosis. These diseases also have an impact on the sensory and motor functioning of patients and may be diagnosed in primary care settings.
 |
ANATOMY, PHYSIOLOGY, AND PATHOLOGY
Oligodendroglia cells of the CNS and Schwann cells of the peripheral nervous system are responsible for forming lipoprotein layers of nerve fibers in early life known as myelin sheaths. There are chemical and immunologic differences between central and peripheral myelin sheaths, but they serve the same function: to promote nerve transmission along the axon. Demyelination, a feature of many neurologic disorders, can result from neuronal damage, as well as damage to the myelin itself, whether it is the result of local injury, ischemia, toxic agents, or metabolic disorders (Berkow, 1992).
Extensive myelin loss is usually followed by axonal degeneration and cell body degeneration and may be irreversible. However, there are instances in which remyelination occurs, and repair, regeneration, and complete recovery of neural function are possible. This is often seen after the segmental demyelination that characterizes many peripheral neuropathies and may explain the exacerbations and remissions of MS. Central demyelination is the predominant finding in several disorders of uncertain etiology that are known as primary demyelinating diseases (Berkow, 1992). The most prominent of these demyelinating diseases is MS, the focus of this chapter.
The pathologic hallmarks of MS are plaques, areas of demyelination restricted to the white matter of the CNS. These sites lack myelin and contain perivascular inflammation. Plaques are frequently found in the white matter of the optic nerves, the brain stem, the cerebellum, and the spinal cord. They are also found in the cerebrum, usually in a perivascular distribution. Occasionally plaques are seen in the gray matter. The location of the plaques correlates with the patient’s symptoms (Lynch & Rose, 1996). The reasons for the patterns of lesion location are not known. However, proximity to the flow of cerebrospinal fluid or vascular perfusion might result in a high concentration of circulating inflammatory proteins in these areas (Sobel, 1995).
Currently, pathologists believe that demyelination is secondary to inflammation. The inflammatory response is perivenular, with a lesser response at the edges of or within plaques. Features of the cellular immune response that may be seen in MS are cells such as mononuclear cells, lymphocytes, and monocytes. These cells are engaged in reactions in the perivascular cuffs, in the leptomeninges, and diffusely in the CNS parenchyma (Sobel, 1995).
Lymphocytes probably contribute to the inflammatory process through antibody and cell-mediated immunity. Secretion of lymphokines and cytokines may be an indirect mechanism (Lynch & Rose, 1996). Cytokines may influence macrophage activation, stimulating the phagocytosis of myelin. Products of the immune response, including immunoglobins, interleukins, interferon, and tumor necrosis factor, accompany the acute MS lesion. After the initial events, immature oligodendrocytes appear and presumably participate in remyelination.
MS lesions may vary in size, the extent of CNS involvement, and the stage of evolution. These lesions may evolve through multiple remyelinating and demyelinating episodes, with the result being a chronic burned-out plaque. An autopsy of a patient with MS may show old inactive plaques, along with actively inflamed lesions and ongoing demyelination (Sobel, 1995).
EPIDEMIOLOGY
There are more than 300,000 people with the diagnosis of MS in the United States. Most cases appear during the third and fourth decades of life, with the risk of the first episode peaking at about age 30 (Vollmer & Waxman, 1991). The incidence of the disease before the age of 10 and after the age of 50 is less than 5%. The risk for development of MS for women is two to three times greater than that for men. The onset of the disease occurs in women at a younger age. The higher incidence
of MS in women may reflect a hormonal factor that increases susceptibility (Matthews, 1991).
of MS in women may reflect a hormonal factor that increases susceptibility (Matthews, 1991).
There appears to be a distinct geographic difference in the incidence and prevalence of MS. In general, these rates escalate with increasing distance from the equator, the highest prevalence being between 40° north and 40° south from the equator. People of higher socioeconomic status are at higher risk, and the relation to urbanization remains unknown (Vollmer & Waxman, 1991).
MS occurs among whites and other ethnic groups, but a higher frequency has been reported in whites than in nonwhites. African Americans are at a lower risk than whites (Sibley et al, 1989). Asians manifest low rates of disease (2 per 100,000 people). There have been no reported cases of MS in the native-born people of Alaska and Greenland (Matthews, 1991).
There has been support for the idea that environment may be related to the development of the disease. In studies on immigrants, it was found that those who moved after age 15 from a high-risk area retained much of the high-risk status of their place of origin. Those moving at a younger age acquired the low-risk status of their new home (Hopkins, 1993). A latent period of approximately 20 years follows before the development of the chronic illness (Vollmer & Waxman, 1991).
Genetic factors have long been recognized to play a role in the development of MS. An increased prevalence of MS in family members of MS patients has been observed. Fifteen percent of patients are known to have an affected relative. Siblings of an affected person are at a higher risk than parents or children (Matthews, 1991).
However, the tendency to consider all diseases with increased familial incidence as hereditary may be erroneous. Virus infection as a possible cause for MS has been studied. Elevated antibody levels to measles are found in MS patients compared with controls. The measles antibody level is also raised in siblings of MS patients. Raised titers of antibodies to other viruses have been reported in cases of MS, including rubella, herpes zoster, herpes simplex, and Epstein-Barr virus (Matthews, 1991).
ETIOLOGY
MS is an immune-mediated disease, acquired by a genetically predisposed persons near the time of puberty. Epidemiologic and pathologic aspects, as well as a host of immunologic and experimental observations, must be taken into account when considering the mechanism of demyelination (Hopkins, 1993). An immune-mediated inflammatory demyelination, probably genetically determined, may be the basis for the disease. Markers for immunopathologic processes, such as perivascular infiltration by lymphocytes, class II major histocompatibility complex
antigen expression by cells in the lesions, lymphokines and cytokines secreted by activated cells, and absence of evidence of infection, are all noted on pathology exams. Other indicators of a possible autoimmune response and theories of autoimmune pathogenesis are listed in Tables 52-2 and 52-3.
antigen expression by cells in the lesions, lymphokines and cytokines secreted by activated cells, and absence of evidence of infection, are all noted on pathology exams. Other indicators of a possible autoimmune response and theories of autoimmune pathogenesis are listed in Tables 52-2 and 52-3.
 |
 |
DIAGNOSTIC CRITERIA
No specific test for MS exists. The evidence supporting diagnosis rests on multiple signs and symptoms and characteristic remissions and exacerbations. MS is characterized by dissemination in time and space—in other words, there are multiple episodes of dysfunction and multiple areas of involvement within the CNS. The diagnosis cannot be made with assurance at the first attack. Table 52-4 lists the criteria for diagnosis.
 |
Course of Disease
Although the course of MS is unpredictable in any given patient, for most patients it is more benign than generally thought. Twenty-five years after diagnosis, an estimated 75% of patients are alive, 66% are ambulatory, and 33% are productively employed (Vollmer & Waxman, 1991). The various courses MS can take are presented in Tables 52-5 and 52-6.
 |
 |
Clinical Symptoms
The initial symptoms may occur alone or in combination. The distribution of pathologic lesions accounts for the wide variety of symptoms. The clinical picture objectifies the loss, permanent or temporary, of normal impulse-conduction properties in myelinated nerve fibers. The fibers can be in the long ascending and descending tracts of the spinal cord, the shorter white matter tracts within the brain stem, or the subcortical white matter of the cerebrum and cerebellum (Vollmer & Waxman, 1991).
The incidence of initial symptoms is as follows: weakness in one or more limbs, 40%; optic neuritis, 22%; paresthesia, 21%; diplopia, 12%; vertigo, 5%; and disturbance of micturition, 5%. There is usually asymmetric distribution of the clinical defects, but as the disease progresses and lesions become more widely distributed, the symptoms become bilateral (Matthews, 1991).
MOTOR SYMPTOMS
Motor symptoms often occur early in the disease. Patients may complain of weakness or heaviness of the involved limb. They may initially complain of weakness on exertion, gradually increasing until it is constantly present. They may describe a tendency to trip or fall or may drag the affected limb (Hickey, 1992). The symptoms may last for minutes to hours and therefore cannot always be observed during an ordinary clinical exam (Matthews, 1991). There may be no abnormal signs on the clinical exam at rest except for the absence of abdominal reflexes, an indication of cortical tract dysfunction. On exercise, weakness, spasticity, and extensor plantar reflexes rapidly appear, only to become reversible at rest (Matthews, 1991). Uhthoff’s phenomenon is a worsening of motor function after a hot shower or a hot bath. This can be of diagnostic significance.
Pyramidal tract involvement produces characteristics of upper neuron disease: spasticity, hyperreflexia, clonus, and extensor plantar responses. Deep tendon reflexes are exaggerated, sustained clonus may be elicited (usually at the ankles), and extensor plantar responses may be observed. Commonly, these signs are asymmetric. Deep tendon reflexes can be decreased if the reflex arc has been interrupted by a lesion at a certain segmental level of the spinal cord. The Achilles reflex may be absent, and sphincter and sexual dysfunction may be present if lesions are found in sacral segments (Gordon et al, 1991).
Triceps jerks are lost more frequently in the upper extremities; ankle jerks are lost more frequently than knee jerks. In established disease in the upper limbs, biceps and supinator jerks may be associated with finger flexion and Hoffman’s sign. Flicking or nipping the nail of the second, third, or fourth finger, if the reflex is present, causes flexion of these fingers and possibly the thumb. Its presence indicates that the tendon reflexes are hyperactive. An increased jaw jerk suggests involvement above the level of the foramen magnum (Matthews, 1991).
Increased muscle tone is an important factor in this disease. In less severe cases, there is spasticity of the extensor muscles of the legs. This occurs in bed at night or when trying to rise in the morning. The legs are held rigidly extended, usually for several minutes. These spasms are seldom painful but are inconvenient. As the disease progresses, the flexor muscles become spastic. The patient may fall without warning while walking. Flexor spasms are frequently painful (Matthews, 1991).
Bilateral involvement of the corticobulbar tracts occurs in some patients; this leads to dysarthria, dysphagia, hyperactive jaw jerks, bifacial paralysis, and apparent emotional lability. Spastic weakness of the muscles of speech is common. In the early stages, speech is often slurred. As the disease progresses, speech becomes explosive or staccato and unintelligible. Scanning speech—slow and measured speech with pauses between syllables—is seen in later stages of the disease (Hickey, 1992).
SENSORY SYMPTOMS
Sensory symptoms are the most common initial features of MS and occur throughout the course of the disease. These symptoms are varied and the onset may be symmetrical. The sensations are described as tingling, numbness, a pins-and-needles sensation, tightness, coldness, or swelling of the limbs or trunk. Complaints of tight bands or straps around the trunk or limbs are characteristic symptoms (Gordon et al, 1991). A common pattern is for unusual sensations to begin in one foot and spread to both lower limbs, the buttock, the perineum, and the trunk within a few days.
The normal sensation of micturition and defecation may be affected, but control remains normal. Vaginal sensation is also diminished. The exam may reveal no loss of light touch or pinprick, but the pinprick may feel distant, as if something is between the pin and the skin. An intensely itching pain, especially in the cervical dermatomes, usually unilateral and occurring in young women, is suggestive of MS (Gordon et al, 1991).
The most frequent sensory signs are varying degrees of impairment of vibration and joint position sense, decrease of pain and light touch in a stocking-and-glove distribution in the four extremities, and patchy areas of reduced pain and light touch perception in the limbs and trunk (Gordon et al, 1991).
Lhermitte’s sign consists of an electric feeling passing down the back to the legs on flexing the neck. This is common in MS but can occur in other conditions (compressive myopathies). The usual course of a sensory episode is toward complete remission within 6 to 8 weeks; for patients who have frequent attacks, these episodes may clear in 10 days. Observant patients may recognize sensory symptoms of a more localized nature that last for only 1 to 2 days, unrelated to exercise, infection, or environmental temperatures (Matthews, 1991). Sensory symptoms tend to remit more frequently than do symptoms involving other systems.
CEREBELLAR INVOLVEMENT
Cerebellar dysfunction is characterized by gait imbalances, difficulty in performing coordinated actions with the arms, and slurred speech. Cerebellar signs are usually uncommon at the
onset of the disease (Matthews, 1991). The early appearance of cerebellar ataxia is an indication of a poor prognosis, and cerebellar signs often persist. There may be difficulty in distinguishing cerebellar signs from those of weakness, spasticity, sensory loss, and vertigo. Cerebellar ataxia naturally affects the gait, but spasticity and weakness are more important causes of disability.
onset of the disease (Matthews, 1991). The early appearance of cerebellar ataxia is an indication of a poor prognosis, and cerebellar signs often persist. There may be difficulty in distinguishing cerebellar signs from those of weakness, spasticity, sensory loss, and vertigo. Cerebellar ataxia naturally affects the gait, but spasticity and weakness are more important causes of disability.
Conventional heel–knee shin tests are poorly performed by weak and spastic legs. Truncal ataxia and gait are not obvious when sitting but contribute to poor balance. These abnormalities may be observed by having the patient stand or walk normally. Cerebellar incoordination in the upper limbs may be terribly disabling, especially when combined with any significant degree of intentional tremor. Severe degrees of intentional tremor may render the hands useless; voluntary movement can be disturbed by violent deviations of the arms that may render the patient unapproachable (Matthews, 1991).
VISUAL INVOLVEMENT/OPTIC NEURITIS
Many patients with MS display abnormalities of vision. These defects are the result of lesions of the optic nerve or chiasm. Even patients who do not demonstrate a clinically detectable visual defect often do so electrophysiologically through the measurement of visual evoked potentials. Evoked potentials are considered extensions of the clinical exam and allow objective measurement of lesions in specific pathways (Rudnick, 1996).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


