Coagulopathies
Ian Rabinowitz M.D.
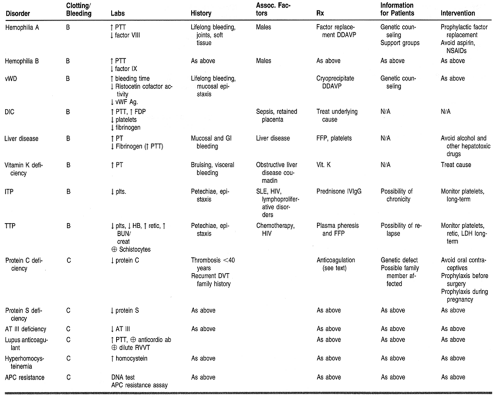
Coagulopathies, include hemorrhage, thrombosis, and embolism and represent common clinical manifestations of hematologic disease. Normally, bleeding is controlled by clot formation, which results from the interaction of platelets, plasma proteins, and the vessel wall. The clot ultimately is dissolved through clot lysis. A derangement of any of these components may result in a bleeding disorder. Individual disease states will be examined under the broad headings of coagulation factor deficiencies, disorders of platelets, and mixed disorders. Thrombotic states will also be examined (Table 37-1).
 |
ANATOMY, PHYSIOLOGY, AND PATHOLOGY
Blood Coagulation
Coagulation is initiated after blood vessels are damaged, enabling the interaction of blood with tissue factor, a protein present beneath the endothelium (Fig. 37-1). Small amounts of factor VII present in plasma bind to tissue factor, and this tissue factor–factor VII complex activates factor X. Activated factor X, in the presence of factor V, activates prothrombin (II) to thrombin (IIa), which subsequently cleaves fibrinogen to fibrin. The fibrin polymerizes into an insoluble gel. This is stabilized by the action of factor XIII. This process constitutes the extrinsic pathway.
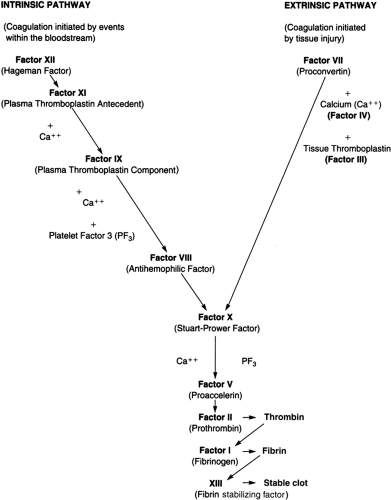 FIGURE 37-1 The coagulation cascade is initiated by the “extrinsic pathway” and consolidated by the “intrinsic pathway.” Coagulation is controlled by the inhibition of factors VIII and V by APC, and by AT III inhibition of II and X. |
Coagulation is consolidated by the intrinsic pathway. Factor XI is activated (possibly by thrombin generated in the extrinsic pathway), resulting in the activation of factor IX, which then activates factor X in the presence of factor VIII. Activated factor X produces a fibrin clot, as outlined above in the extrinsic pathway. Decreased levels of clotting factors may be caused by defective synthesis, excessive use, circulating inhibitors of clotting factors, or excessive proteolysis by the fibrinolytic system.
The coagulation pathway is controlled by a number of mechanisms. Protein C is a plasma protein that inhibits factors V and VIII, thus inhibiting the activity of factors IX and X, respectively. This inhibition is catalyzed by protein S. Antithrombin III (AT III) primarily inhibits the activity of thrombin and factor X, and this inhibition is greatly enhanced by heparin. Loss of function of these proteins would result in uninhibited coagulation and hence a predisposition to spontaneous thrombosis.
Fibrinolysis is a mechanism for dissolving fibrin clots. Plasmin, the activated form of plasminogen, cleaves fibrin to produce soluble fragments. Fibrinolytics such as tissue plasminogen activator and urokinase activate plasminogen, resulting in dissolution of a fibrin clot.
CLASSES OF BLEEDING DISORDERS
Vascular Defects
Vascular defects usually cause bleeding only into the skin and mucous membranes. Congenital causes include Osler-Weber-Rendu syndrome and Ehlers-Danlos disease. Acquired causes of vascular purpura include infections, drugs, uremia, connective tissue disorders, and dysproteinemias. Treatment is directed to the primary illness.
Inherited Coagulation Disorders
Deficiencies of any of the known coagulation factors are present from birth and may be inherited or result from a spontaneous disruption in the associated coagulation factor gene. Only deficiencies of factors VIII and IX, and von Willebrand’s disease (vWD) will be discussed in more detail, because the other disorders are rare.
HEMOPHILIA
Hemophilia is an X-linked recessive disorder associated with a congenital deficiency of factor VIII or IX (hemophilia A or B, respectively). Hemophilia is suspected in any male who has excessive bleeding after trauma, or spontaneous bleeding into joints or soft tissues. Patients with severe hemophilia (factor level <1%) are at risk for spontaneous hemarthrosis and soft-tissue bleeding. Patients who have moderate disease (factor level 1% to 4%) or mild hemophilia (factor level 5% to 25%) are at a reduced risk of spontaneous hemorrhage, but may bleed excessively after trauma or surgery.
VON WILLEBRAND’S DISEASE
von Willebrand factor (vWF) is a plasma protein that is required for the adhesion of platelets to sites of vascular damage. Deficiency of vWF is called type I; qualitative abnormality of vWF is called type II. vWD is an autosomally inherited hemostatic disorder of variable severity, characterized by mucosal and cutaneous bleeding, similar to patients with platelet disorders. Patients may have a prolonged bleeding time and a prolonged activated partial thromboplastin time (aPTT).
Mixed Coagulation Disorders
Mixed coagulation disorders are a group of acquired diseases. They usually involve multiple elements of the hemostatic system and are often associated with a particular disease or clinical syndrome.
DISSEMINATED INTRAVASCULAR COAGULATION
Disseminated intravascular coagulation (DIC) can be caused by the activation of either the coagulation or fibrinolytic system and therefore can result in bleeding or thrombosis. Conditions associated with DIC include infections such as gram-negative sepsis, meningococcemia, Rocky Mountain spotted fever, and typhoid fever; obstetric complications (abruptio placentae, eclampsia, retained dead fetus); massive trauma; surgery; shock; and certain malignancies.
LIVER DISEASE
Coagulation disorders are common in patients with liver disease. These may arise from malabsorption of vitamin K, decreased synthesis of clotting proteins, or synthesis of abnormal proteins. Fibrin degradation products may be elevated because of poor hepatic clearance, resulting in impaired coagulation resembling DIC. Liver disease associated with hypersplenism may result in thrombocytopenia.
VITAMIN K DEFICIENCY
Factors II, VII, IX, and X are vitamin K-dependent coagulation factors that are synthesized in the liver. Vitamin K is naturally synthesized by intestinal bacteria. It is a fat-soluble vitamin and is absorbed only in the presence of bile salts. Depletion of vitamin K-dependent factors may occur in patients receiving antibiotics; in obstructive jaundice, malabsorptive states, or hepatic parenchymal disease; and in patients receiving warfarin therapy. Patients have an increased, international normalization ratio (INR).
Platelet Disorders
Platelet disorders include thrombocytopenia, which may be caused by diminished platelet production, enhanced platelet destruction, or sequestration of platelets (Table 37-2). Qualitative platelet disorders may be congenital or acquired. Congenital disorders may affect platelet adhesion, aggregation, or platelet secretion, and are rare. Acquired qualitative platelet disorders are secondary to uremia, myeloproliferative disorders, drugs, and dysproteinemias.
 |
IMMUNE THROMBOCYTOPENIC PURPURA
Immune thrombocytopenic purpura (ITP), the autoimmune destruction of platelets, usually presents as ecchymoses, petechiae, or bleeding. The differential diagnosis includes aplastic
anemia, sepsis, DIC, acute leukemia, drug-induced thrombocytopenia, and infiltrative bone marrow disorders. The diagnosis is made by excluding other causes of thrombocytopenia, after a careful history, physical exam, and peripheral smear. Patients with risk factors for HIV should be tested for the HIV antibody. Bone marrow aspiration may be indicated in elderly patients or patients with atypical findings.
anemia, sepsis, DIC, acute leukemia, drug-induced thrombocytopenia, and infiltrative bone marrow disorders. The diagnosis is made by excluding other causes of thrombocytopenia, after a careful history, physical exam, and peripheral smear. Patients with risk factors for HIV should be tested for the HIV antibody. Bone marrow aspiration may be indicated in elderly patients or patients with atypical findings.
Chronic ITP usually occurs in adults, with a 3:1 ratio of females to males. It is common in patients between the ages of 20 and 50 years. ITP may also be associated with HIV infection, systemic lupus erythematosus, lymphoproliferative disorders, ulcerative colitis, and carcinoma. The thrombocytopenia is often chronic and unremitting, requiring definitive therapy. Acute ITP occurs primarily in children.
DRUG-INDUCED THROMBOCYTOPENIA
Drug-induced thrombocytopenia is diagnosed after excluding other causes, and by noting a temporal relation between the onset of thrombocytopenia and the administration of the drug, as well as resolution on discontinuation of the drug. Although any drug can be implicated, alcohol, thiazide diuretics, quinine, quinidine, penicillins, gold, sulfa, and heparin are more commonly associated with drug-induced thrombocytopenia. Myelosuppressive drugs used to treat malignancies and other disorders can produce thrombocytopenia by suppressing platelet production.
Heparin-induced thrombocytopenia is associated with an early nonimmune clinical syndrome and a later immune-mediated thrombocytopenia that occurs 5 to 7 days after the initiation of heparin. This type of thrombocytopenia may be associated with paradoxical thrombosis instead of bleeding.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


