CHAPTER 17 Cardiogenic Shock
DRAMATIC ADVANCES during the past several decades in diagnosing, monitoring, and treating patients with acute myocardial infarction (MI) have decreased hospital mortality rates by 50%. The organization of coronary care units in the 1960s to treat lethal arrhythmias1 and the development of fibrinolytic therapy in the 1980s to reduce infarct size2–5 were the biggest breakthroughs. Cardiogenic shock, not arrhythmia, is now the most common cause of death in patients hospitalized with acute MI. However, neither the incidence nor the mortality rate associated with cardiogenic shock has been reduced by modern cardiac intensive care unit interventions, including vasopressor and inotropic drug infusions, hemodynamic monitoring, and intra-aortic balloon pump (IABP) counterpulsation (Table 17-1).6–12 More recent reports show a survival advantage, however, for patients who undergo successful reperfusion with percutaneous coronary intervention (PCI) or coronary artery bypass graft (CABG) surgery.13–19 This chapter reviews the epidemiology, pathogenesis, clinical presentation, and current management of cardiogenic shock.
Table 17–1 Historical Milestones in Cardiogenic Shock
| 1934 | Fishberg et al6 described the shock state as a peripheral complication of myocardial infarction |
| 1942 | Stead and Ebert7 attributed the shock state to extreme myocardial dysfunction |
| 1954 | Griffith et al8 used L-norepinephrine as pressor support |
| 1967 | Killip and Kimball1 showed no survival advantage with coronary care unit monitoring |
| 1968 | Kantrowitz et al9 described the clinical use of the IABP |
| 1972 | Dunkman et al10 showed successful treatment with CABG surgery |
| 1973 | Scheidt et al11 showed no survival advantage with IABP |
| 1976 | Forrester et al12 defined hemodynamic subsets using the pulmonary artery catheter |
| 1980 | DeWood et al13 showed a survival advantage with early CABG surgery |
| 1980 | Mathey et al14 showed successful treatment with fibrinolytic therapy |
| 1982 | Meyer et al15 showed successful treatment with PTCA |
| 1988 | Lee et al16 showed a survival advantage with PTCA |
| 1999 | Hochman et al17–19 proved a survival advantage with revascularization in the SHOCK trial |
CABG, coronary artery bypass graft; IABP, intra-aortic balloon pump; PTCA, percutaneous transluminal coronary angioplasty.
Epidemiology
Definition
The diagnosis of cardiogenic shock should include the following:
Etiology
The most common cause of cardiogenic shock is acute MI.20 Often, anterior MI from acute thrombotic occlusion of the left anterior descending artery results in extensive infarction. Alternatively, a smaller MI in a patient with borderline left ventricular function may be responsible for insufficient cardiac output. Large areas of ischemic nonfunctioning but viable myocardium occasionally lead to shock in patients with MI. The delayed onset of shock may result from reocclusion of a patent infarct artery, infarct extension, or metabolic decompensation of non–infarct zone regional wall motion. Occasionally, right ventricular MI from occlusion of a proximal large right coronary artery in a patient with inferior MI is the cause.21
Mechanical complications unrelated to infarct size account for approximately 12% of cases. The papillary muscle of the mitral valve may infarct or rupture, causing acute, severe mitral regurgitation.22 Rupture of the interventricular septum causing ventricular septal defect23 or rupture of the left ventricular free wall producing pericardial tamponade24 also needs to be considered. Other causes of cardiogenic shock that are not emphasized in this chapter include end-stage cardiomyopathy, myocardial contusion, myocarditis, hypertrophic cardiomyopathy, valvular heart disease, pericardial disease, and post–cardiopulmonary bypass.
Incidence
Before the emphasis on time-to-treatment and primary PCI, the incidence of cardiogenic shock had remained unchanged for more than 25 years with approximately 8% of patients with ST segment elevation myocardial infarction (STEMI)25,26 and 2.5% of patients with non–ST segment elevation MI27,28 developing cardiogenic shock. The latter group is more likely to have circumflex artery occlusion, comorbid disease, and severe three-vessel disease or left main disease.28 Cardiogenic shock usually develops early after onset of symptoms, with approximately half of patients developing shock within 6 hours and 72% developing shock within 24 hours.29 Others first develop a preshock state manifested by systemic hypoperfusion without hypotension.30 These patients benefit from aggressive supportive therapy, and revascularization; early intervention may abort the onset of cardiogenic shock.
Pathogenesis
Pathology
The early development of cardiogenic shock is usually caused by acute thrombosis of a coronary artery supplying a large myocardial distribution, with no collateral flow recruitment.31 Frequently, this is the left anterior descending artery, although shock may result from coronary thrombosis in other sites if previous MI has occurred. Multivessel disease is present in two thirds of patients.32
Autopsy studies have consistently shown that at least 40% of the myocardium is infarcted in patients who die of cardiogenic shock.33 Various ages of infarction reflect previous infarction, reinfarction, or infarct extension.
The infarct border zone in patients without hypotension is clearly demarcated. In patients dying of shock, it is irregular, with marginal extension. Focal areas of necrosis remote from the infarct zone are also present. These findings result from progressive cell death owing to poor coronary perfusion, are reflected by prolonged release of cardiac enzymes, and contribute to hemodynamic deterioration.34
Pathophysiology
Progressive hemodynamic deterioration leading to cardiogenic shock results from a sequence of events (Fig. 17-1). A critical amount of ischemic or necrotic myocardium decreases contractile mass and cardiac output. When cardiac output is low enough that arterial blood pressure declines, coronary perfusion pressure decreases in the setting of an elevated left ventricular end-diastolic pressure. The resulting reduction in coronary perfusion pressure gradient from epicardium to endocardium exacerbates myocardial ischemia, further decreasing left ventricular function and cardiac output, perpetuating a vicious cycle. The speed with which this process develops is modified by the infarct zone, remote myocardial function, neurohumoral responses, and metabolic abnormalities.
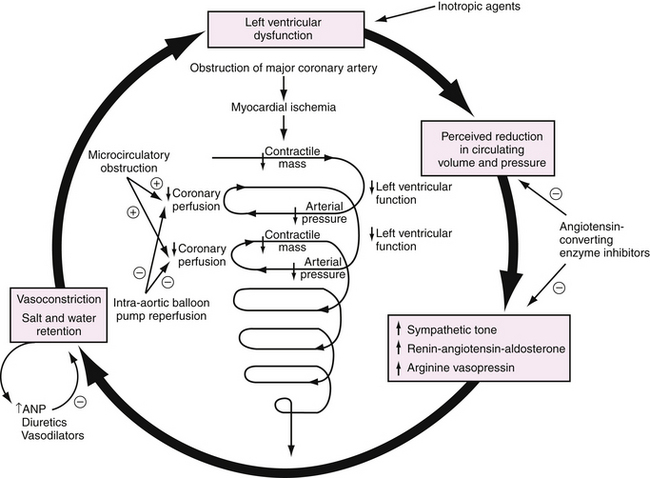
Preclinical and clinical studies35 have shown the importance of hypercontractility of remote myocardial segments in maintaining cardiac output in the setting of a large MI. This compensatory mechanism is lost when multivessel disease is present and produces ischemia in noninfarct segments.
A systemic inflammatory state with high plasma levels of cytokines (e.g., tumor necrosis factor-α, interleukin-6) and inappropriate nitric oxide production additionally may depress myocardial function or impair catecholamine-induced vasoconstriction. All of these factors lead to diminished coronary artery perfusion and trigger a vicious cycle of further myocardial ischemia and necrosis resulting in even lower blood pressure, lactic acidosis, multiple organ failure, and ultimately death.36
Clinical Presentation
History and Physical Examination
The diagnosis of acute MI must be confirmed. Noncardiac causes of shock need to be ruled out, including aortic dissection, tension pneumothorax, massive pulmonary embolism, ruptured viscus, bleeding, and sepsis. Risk factors for developing cardiogenic shock include older age, anterior MI location, hypertension, diabetes mellitus, multivessel coronary artery disease, prior MI, prior congestive heart failure, STEMI, or left bundle branch block.37,38
Echocardiography
Echocardiography can be performed rapidly and offers valuable information on the extent of left ventricular dysfunction. A dilated, hypokinetic left ventricle suggests left ventricular shock, whereas a dilated right ventricle suggests right ventricular involvement. Normal ventricular function, low cardiac output, and mitral regurgitation are consistent with acute severe mitral regurgitation. Pericardial tamponade from hemorrhagic effusion or free wall rupture can be detected quickly. The Doppler evaluation can easily confirm the presence of significant mitral regurgitation or ventricular septal rupture. Transesophageal echocardiography is helpful in patients in whom image quality is inadequate, or when a flail mitral leaflet is suspected but not seen on transthoracic echocardiography.
Management
General Measures
Numerous supportive measures need to be instituted quickly (Fig. 17-2). If there is no clinical evidence for pulmonary edema, a fluid bolus should be given to exclude hypovolemia as a cause of hypotension. Patients with a history of inadequate fluid intake, diaphoresis, diarrhea, vomiting, or diuretic use may not have pump failure and improve dramatically with fluid administration. Because preload is critical in patients with right ventricular shock, fluid support and avoidance of nitrates and morphine are indicated (Table 17-2).
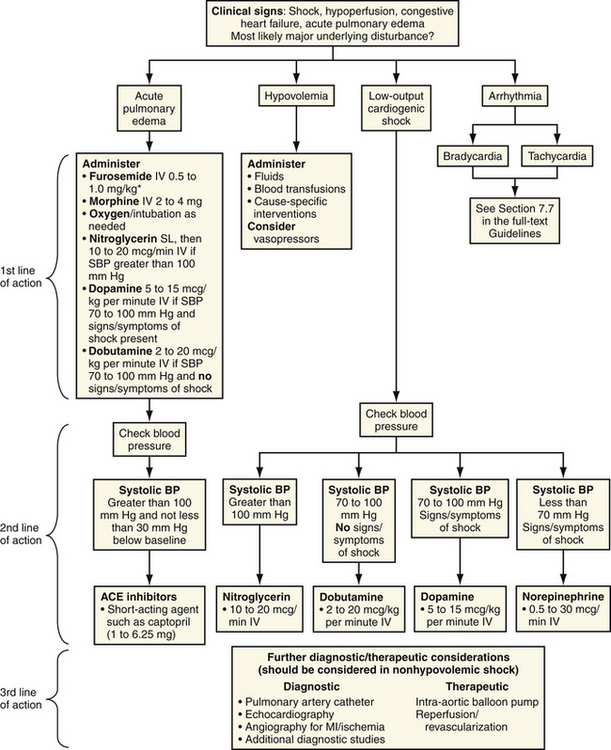
Table 17–2 Conventional Therapy for Cardiogenic Shock
| 1. Maximize volume (RAP 10-14 mm Hg, PAWP 18-20 mm Hg) |
| 2. Maximize oxygenation (e.g., ventilator) |
| 3. Correct electrolyte and acid-base imbalances |
| 4. Control rhythm (e.g., pacemaker, cardioversion) |
| 5. Sympathomimetic amines (e.g., dobutamine, dopamine, norepinephrine) |
| 6. Phosphodiesterase inhibitors (e.g., milrinone) |
| 7. Vasodilators (e.g., nitroglycerin, nitroprusside) |
| 8. Intra-aortic balloon counterpulsation |
PAWP, pulmonary artery wedge pressure; RAP, right atrial pressure.
Hemodynamic Monitoring
Central hemodynamic monitoring is crucial for confirming the diagnosis and guiding pharmacologic therapy (Table 17-3). Urine output needs to be monitored hourly through catheter drainage. An arterial catheter allows constant monitoring of the blood pressure. A pulmonary artery catheter should be inserted as soon as feasible to measure intracardiac pressures, cardiac output, systemic resistance, and mixed venous oxygen saturation. Although use of the pulmonary artery catheter has not been associated with mortality benefit in patients without MI, it is very helpful in the titration of fluids and medications in patients with cardiogenic shock.
Table 17–3 Hemodynamic Profiles
| Left ventricular shock | High PCWP, low CO, high SVR |
| Right ventricular shock | High RA, RA/PCWP >0.8, exaggerated RA y descent, RV square root sign |
| Mitral regurgitation | Large PCWP v wave |
| Ventricular septal defect | Large PCWP v wave, oxygen saturation step-up (>5%) from RA to RV |
| Pericardial tamponade | Equalization of diastolic pressures approximately 20 mm Hg |
CO, cardiac output; PCWP, pulmonary capillary wedge pressure; RA, right atrium; RV, right ventricle; SVR, systemic vascular resistance.
The hemodynamic profile of left ventricular shock, as defined by Forrester and coworkers,12 includes pulmonary artery wedge pressure greater than 18 mm Hg and a cardiac index less than 2.2 L/min/m2. Others have used a pulmonary wedge pressure of 15 mm Hg or 12 mm Hg and a cardiac index of 2 L/min/m2 or 1.8 L/min/m2. The hemodynamic profile of right ventricular shock includes right atrial pressure of 85% or more of the pulmonary artery wedge pressure, steep y descent in the right atrial pressure tracing, and the dip and plateau (i.e., square root sign) in the right ventricular waveform. Large v waves in the pulmonary artery wedge tracing suggest the presence of severe mitral regurgitation. An oxygen saturation step-up (>5%) from the right atrium to the right ventricle confirms the diagnosis of ventricular septal rupture. Equalization of right atrial, right ventricular end-diastolic, pulmonary artery diastolic, and pulmonary capillary wedge pressures occurs with severe right ventricular infarction or pericardial tamponade owing to free wall rupture or hemorrhagic effusion. Cardiac power (mean arterial pressure × cardiac output/451) is the strongest hemodynamic predictor of hospital mortality.39
Pharmacologic Support
Vasopressor and inotropic drug support are the major initial interventions for reversing hypotension and improving vital organ perfusion (Table 17-4). Failure to improve blood pressure with these agents is an ominous prognostic sign. Continued hypotension results in progressive myocardial ischemia and deterioration of ventricular function. Although many patients temporarily respond to therapy, hospital mortality rates remain unchanged without successful reperfusion therapy.
Table 17–4 Pharmacologic Treatment for Cardiogenic Shock
| Drug | Doses | Side Effects |
|---|---|---|
| Dobutamine | 5-15 μg/kg/min IV | Tolerance |
| Dopamine | 2-20 μg/kg/min IV | Increased oxygen demand |
| Norepinephrine | 0.5-30 μg/min IV | Peripheral and visceral vasoconstriction |
| Nitroglycerin | 10 μg/min, increased by 10 μg every 10 min, maximum200 μg/min IV | Headache, hypotension, tolerance |
| Nitroprusside | 0.3-10 μg/min IV | Hypotension, cyanide toxicity |
| Milrinone | 50 μg/kg over 10 min IV, then 0.375-0.75 μg/kg/min | Ventricular arrhythmia |
| Furosemide | 20-160 mg IV | Hypokalemia, hypomagnesemia |
| Bumetanide | 1-3 mg IV | Nausea, cramps |
IV, intravenously.
Catecholamine infusions should be carefully titrated. A delicate balance must be obtained between increasing coronary perfusion pressure and increasing oxygen demand, so that myocardial ischemia is not exacerbated. Excessive peripheral vasoconstriction decreases tissue perfusion, increased afterload increases filling pressures, and excessive tachycardia or arrhythmias can be stimulated. Extravasation of dopamine or norepinephrine can cause tissue necrosis.
Related posts:
 Evolution of the Coronary Care Unit: Past, Present, and Future
Evolution of the Coronary Care Unit: Past, Present, and Future
 Conduction Disturbances in Acute Myocardial Infarction
Conduction Disturbances in Acute Myocardial Infarction
 Diagnosis and Treatment of Complications of Coronary and Valvular Interventions
Diagnosis and Treatment of Complications of Coronary and Valvular Interventions
 Pharmacologic Interactions in the CICU
Pharmacologic Interactions in the CICU
 Acute Aortic Syndromes: Diagnosis and Management
Acute Aortic Syndromes: Diagnosis and Management
 Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


