CHAPTER 37 Antiplatelet Therapy
The Platelet and Acute Coronary Syndromes
Plaque rupture and luminal thrombus formation are the sentinel events that convert the atherosclerotic disease process from a slowly progressive condition causing insidious luminal obstruction to an acute coronary event that may lead to clinical deterioration and possible death. Plaque rupture is a term that describes the development of a gap in the fibrous cap exposing its collagen and the underlying lipid-rich core to flowing blood.1 This event results in activation of the circulating platelets, which in turn initiates a sequence of events involving the activated platelet: adhesion, aggregation, and secretion (Fig. 37-1).
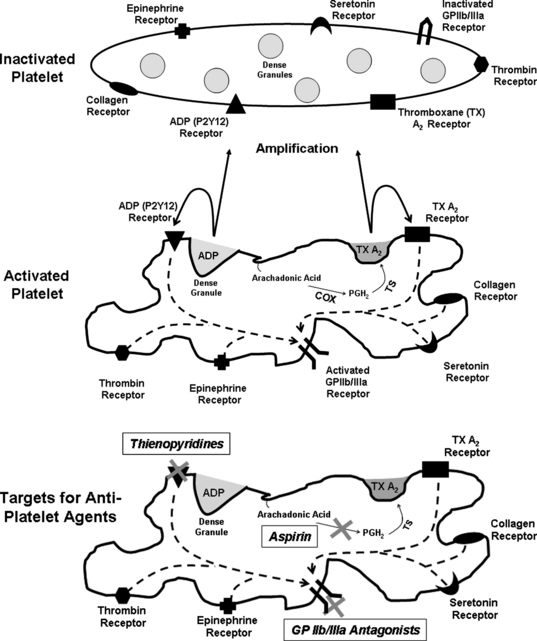
Platelet activation occurs via multiple stimuli including adenosine diphosphate (ADP) and thromboxane A2, but collagen and thrombin are the two most potent platelet activators. Once collagen is available for binding, platelet adhesion and activation depend on two major collagen receptors located on the platelet membrane: the integrin glycoprotein Ia/IIa (α2ß1) and glycoprotein VI. Both of these receptors generate activation signals that enhance platelet thrombus formation. Thrombin is also a potent stimulus for platelet activation by binding and cleaving platelet protease activated receptors (PAR), PAR1, and PAR4. This process forms a tethered-ligand that acts to initiate transmembrane signaling, also leading to platelet activation.2–4
Once activated, platelet adhesion occurs when platelet glycoprotein Ib-IX-V binds to tissue von Willebrand factor (vWF).5 At least one other receptor that participates in platelet adhesion includes the platelet receptor Ia/IIa, which binds to collagen fibrils.6 Platelet aggregation soon follows and is primarily mediated by the glycoprotein IIb/IIIa complex (αIIbß3). Glycoprotein IIb/IIIa adhesion proteins are found on platelets and megakaryocytes. They are the most abundant glycoprotein found on the platelet surface with approximately 80,000 copies. The glycoprotein IIb/IIIa complex requires platelet activation, which causes a conformational change in the receptor thereby becoming a high-affinity fibrinogen binding receptor. Fibrinogen bridges activated platelets forming a platelet plug. The cytosolic portion of the glycoprotein IIb/IIIa complex stimulates actin rearrangement resulting in platelet spreading, aggregation, and clot retraction. Glycoprotein IIb/IIIa receptor antagonists are powerful antiplatelet drugs and will be discussed in detail later in this chapter.2–4
Activated platelets secrete substances from their granules, which stimulate further activation and aggregation. These mediators include, but are not limited to, adenosine diphosphate (ADP), thromboxane A2, fibrinogen, fibronectin, and thrombospondin. There are at least two receptors that bind ADP: P2Y12 and P2Y1. The thienopyridine drugs prasugrel, clopidogrel, and ticlopidine block the P2Y12 receptor. Thromboxane A2 is an arachidonic-acid metabolite and a strong platelet stimulator. The production of thromboxane A2 depends on the cyclo-oxygenase pathway, which is irreversibly inhibited by acetyl salicylic acid or aspirin (see Fig. 37-1).2–4 It should be noted that the aforementioned steps of adhesion, aggregation, and secretion do not occur in a stepwise fashion, but are presented that way for simplicity. In reality, these processes occur simultaneously and much work is still needed to better define targets for future antiplatelet drugs.
Antiplatelet Therapy for Unstable Angina and Non-STEMI
As previously described, the first step in the process of coronary thrombus formation involves platelet activation and aggregation. Therefore antiplatelet therapy is the cornerstone of treatment in UA and NSTEMI. Numerous clinical trials have established that appropriate antiplatelet therapy in UA and NSTEMI reduces mortality and other ischemic events. When antiplatelet therapy is used in the coronary care unit (CCU), it is imperative that physicians ensure their patients are receiving these agents at the appropriate dosages and cautiously watch for side effects, namely bleeding and hematologic abnormalities.
Acetyl Salicylic Acid (ASA or Aspirin)
When aspirin is given acutely as a tablet, it should be chewed to achieve rapid platelet inhibition within approximately 20 minutes. Aspirin blocks the production of thromboxane A2 by irreversibly acetylating a serine residue on cyclo-oxygenase 1. This prevents the conversion of arachidonic acid to prostaglandin-H2, a precursor of thromboxane A2. Thromboxane A2 causes vasoconstriction and platelet aggregation.7 Unless there is a well-documented severe allergic reaction, all patients with acute coronary syndrome should be treated with aspirin.
The benefit of aspirin in unstable angina has been demonstrated in several trials. In the Veterans Administration Cooperative Trial, 1266 men with unstable angina were randomized to receive either aspirin or placebo. The primary end points were death and myocardial infarction (MI). The incidence of death or acute MI was reduced by 51% in the aspirin group (5.0% versus 10.1%, p = 0.0005). Examined individually, both nonfatal acute MI and death were dramatically reduced by more than 50% in the aspirin group, although the reduction in death did not quite reach the level of statistical significance (3.4% versus 6.9%, p = 0.005 for acute MI and 1.6% versus 3.3%, p = 0.054).8 Further conformation for the benefit of aspirin was provided in the Canadian multicenter trial where 555 unstable angina patients were randomized to aspirin, sulfinpyrazone, both, or neither. In those randomized to aspirin, there was a significant risk reduction of 51% in combined end points of death and nonfatal MI (p = 0.008).9 Individual end points of both death and nonfatal myocardial infarction reached statistical significance. Sulfinpyrazone showed no clinical benefit in this trial. Further trials in patients with unstable angina continued to demonstrate a significant reduction in event rates in those receiving aspirin therapy.10
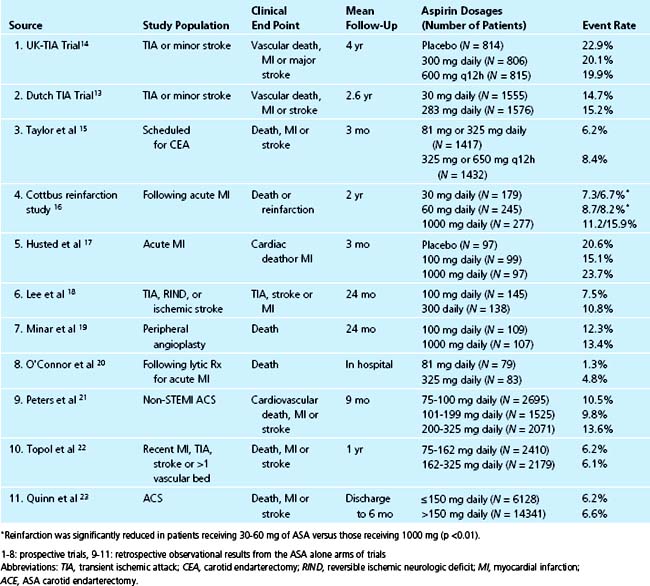
Despite the extensive body of literature describing the benefits of aspirin, little is known about the most effective dose. As little as 30 mg/day chronically can completely inhibit serum TXB2 production,11 and many studies suggest equal benefit with doses less than 150 mg daily compared to higher doses when taken long-term (Fig. 37-2).12 For instance, in 3131 patients with minor stroke or CVA, 30 mg of aspirin daily was just as effective as 283 mg daily in preventing death from vascular causes, nonfatal stroke, or nonfatal myocardial infarction.13 Many large, blinded, controlled trials in addition to several meta-analyses of placebo-controlled trials have evaluated the optimal aspirin dose in treating patients with nearly every clinical manifestation of atherosclerosis including stroke, transient ischemic attack (TIA), percutaneous coronary and peripheral interventions, carotid endarterectomy, and myocardial infarctions. In all of these trials, there is no relationship between increasing aspirin dosage and improved clinical efficacy12 (Table 37-1, see Fig. 37-2).
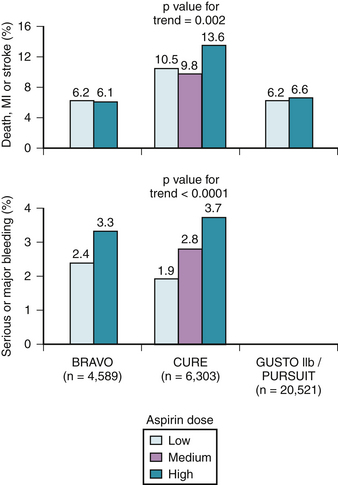
Although only 30 mg/day of aspirin is needed to inactivate thromboxane production, the rapidity of platelet inactivation should also be considered in the context of ACS. In a small study involving 18 healthy volunteers, chewing 162 mg and 324 mg of aspirin resulted in maximal inhibition of TXA2 within 15 minutes. This was not seen with the 81 mg dose.24 When comparing 162 mg aspirin with 325 mg aspirin, both doses seem equally effective in lowering mortality in patients with an STEMI, however, recent data suggests that the 162 mg dose is associated with fewer episodes of bleeding. This was noted in a recent study where acute mortality and bleeding risk was evaluated in fibrinolytic-treated STEMI patients. Data was obtained from the GUSTO I and GUSTO III trials. After adjustment, aspirin dose (162 mg versus 325 mg) was not associated with 24-hour, 7-day, or 30-day mortality rates. There was also no noted difference in MI between the two groups. However, in-hospital moderate/severe bleeding occurred in 9.3% of those treated with 325 mg versus 12.2% among those receiving 162 mg (p <0.001). After adjustment, 325 mg was associated with a significant increase in moderate/severe bleeding (OR, 1.14; 95% CI, 1.05 to 1.24; p = 0.003).25
To assess the effect of aspirin dosage on number and severity of bleeding episodes, data from 31 trials including 192,036 patients was analyzed. The dosage of aspirin was compared with bleeding complications and these data were divided into several categories, including major bleeding, minor bleeding, gastrointestinal, stroke, fatal/life-threatening, and total bleeding episodes. In all of the aforementioned categories, aspirin doses higher than 100 mg/day were associated with significantly more events.26
The ACC/AHA guidelines for treatment of UA/NSTEMI give aspirin a class IA recommendation for immediate and indefinite use in this patient population.27 According to these recommendations, 162 to 325 mg should be administered once ACS is suspected. In patients who do not receive a stent, the first dose should be followed by 75 to 162 mg/day indefinitely. The same daily aspirin dose is recommended in patients who receive stents except during months following stent placement where the aspirin dose should be higher. In patients receiving a bare metal stent, 162 to 325 mg/day is recommended for the first month after stenting. In patients who receive a drug eluting stent, 162 to 325 mg/day for 3 to 6 months is recommended.
Adenosine Diphosphate Receptor Antagonists
Clopidogrel
The introduction of clopidogrel to the field of ACS management has had a dramatic effect on the algorithms of therapy used all over the world. Clopidogrel has been found to be as effective, if not more effective, than aspirin when used in secondary prevention of vascular events. This was demonstrated in the CAPRIE trial where 19,185 patients with recent ischemic stroke, recent MI, or symptomatic peripheral arterial disease were randomized to aspirin (325 mg) or clopidogrel (75 mg) and followed for 1 to 3 years. Patients assigned to receive clopidogrel had a statistically significant relative-risk reduction in the composite outcome of ischemic stroke, MI, or vascular death when compared with aspirin.28 There was no significant difference in side effects between the two treatment groups.
The typical daily dose of clopidogrel is 75 mg daily. In circumstances when rapid platelet inhibition is desired such as ACS, a loading dose of clopidogrel at 300 or 600 mg can be given and has demonstrated additional antiplatelet aggregation properties when compared with aspirin alone.29,30
When added to aspirin in patients with UA/NSTEMI, clopidogrel offers additional reduction in vascular events and death, as established in the CURE study.31 The study randomized 12,562 patients having an NSTEMI (defined by clinical symptoms plus elevated cardiac enzymes or electrocardiographic changes) to receive aspirin plus clopidogrel versus aspirin plus a placebo. The study was designed to examine two primary outcomes: the first was a composite of cardiovascular death, MI, or stroke and the second consisted of the first primary outcome plus refractory ischemia. At 9 months, patients receiving aspirin plus clopidogrel had a statistically significant reduction in the first primary outcome (9.3% versus 11.4%, p <0.001) (Fig. 37-3). Similarly, clopidogrel was associated with a statistically significant reduction in the second primary outcome (16.5% versus 18.8%, p <0.001).31
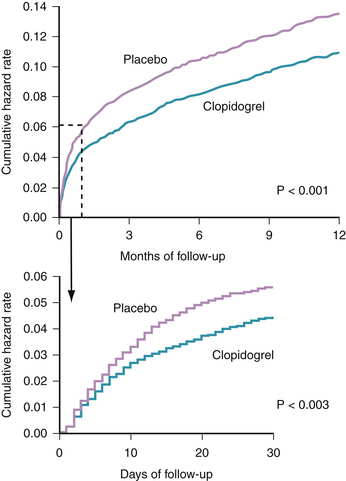
Several observations solidified the role of clopidogrel in management of this patient population. The benefit appeared within 24 hours of presentation and was seen irrespective of the baseline risk stratification.32 Additionally, the continuation of clopidogrel after hospitalization in this group of patients further reduced subsequent ischemic vascular events (cardiovascular death, MI, or stroke). Moreover, the reduction in vascular events was independent of concomitant use of antiplatelet and antithrombotic medication, antihypertensives, lipid lowering therapy, or coronary intervention.33 However, a statistically significant increase in bleeding (based on the CURE definition) was seen in patients on dual antiplatelet therapy.31
With most patients with NSTEMI or severe UA being referred to coronary angiography and revascularization early in the course of their admission to the CCU, it is important to clearly define the value of clopidogrel therapy in patients undergoing PCI. The PCI CURE substudy tested the hypothesis that treatment with clopidogrel plus aspirin, before PCI would reduce post-procedure ischemic complications. Additionally, this trial evaluated the efficacy of long-term clopidogrel use along with aspirin in further reducing vascular complications after PCI. Patients were pretreated with aspirin and either clopidogrel or placebo for a median of 6 days before PCI. After stenting, more than 80% of patients were started on open label ticlopidine or clopidogrel for 4 weeks, and then resumed taking placebo or clopidogrel for an additional 8 months. In the clopidogrel arm, the primary outcome of death, MI, or urgent target vessel revascularization within 30 days was significantly reduced (4.5% versus 6.4% in the aspirin alone arm, p = 0.03). In patients receiving clopidogrel for at least 8 months, there were fewer events of cardiovascular death, MI, or revascularization from any cause (p = 0.03).34
A limitation of PCI CURE was that patients were treated with clopidogrel for an average of 6 days before PCI. With the more rapid and early invasive strategies for managing NSTEMI and UA patients in the United States, this pretreatment duration is logistically unrealistic. The CREDO trial assessed the utility of clopidogrel loading less than 24 hours before PCI. In this trial, 2116 patients undergoing elective PCI for symptomatic coronary artery disease were randomized to receive clopidogrel 300 mg loading dose or placebo 3 to 24 hours before PCI. Subsequent to PCI, all patients received clopidogrel for an additional 28 days. From day 29 to 12 months, those individuals in the control group received a placebo while those loaded on clopidogrel received an additional 75 mg/day of clopidogrel for 12 months. At 28 days only a nonsignificant trend toward benefit was found with pretreatment with a loading dose compared with no pretreatment and no loading dose. However, in a post hoc analysis, patients who received the clopidogrel loading dose between 6 to 24 hours before PCI had a nearly significant 38.6% relative risk reduction in the composite of death, MI, or urgent target vessel revascularization (p = 0.051).35 Further examination of this data revealed that a statistically significant reduction in death, MI, or urgent target vessel revascularization does not actually occur with the 300 mg loading dose unless it is given at least 15 hours before PCI.36
The 600 mg dose has recently been found to inhibit platelets more rapidly and effectively than the 300 mg dose.37–39 Clinically, this may translate into fewer short-term ischemic events after PCI. Recently, Cuisset and colleagues demonstrated that patients who received the 600 mg loading dose of clopidogrel before PCI for an NSTEMI had fewer subsequent ischemic events (defined as cardiovascular death, acute or subacute stent thrombosis, recurrent ACS, and stroke) within 1 month, when compared to those receiving a 300 mg loading dose (5% versus 12%; P = 0.02).38 These findings were subsequently confirmed in another trial: the ARMYDA-2. In that study, 255 patients scheduled for PCI were randomized to either a 300 mg or 600 mg loading dose of clopidogrel 4 to 8 hours before the procedure. The primary end point was 30-day death, MI, or target vessel revascularization. Levels of troponin, CK, and CK-MB were measured at baseline, 8 and 24 hours after the intervention. The primary end point was reached in 4% of patients receiving 600 mg and 12% in patients receiving 300 mg clopidogrel (p = 0.041). The benefit of the higher loading dose was due to a reduction in periprocedural MI. There was also a significant reduction in peak values of troponin, CK, and CK-MB (P = 0.038).40
The AHA/ACC guidelines for management of ACS give clopidogrel a class IA indication in patients who cannot take aspirin. If an early invasive strategy is taken, a preprocedural load and maintenance dose of clopidogrel should be given (IA). If an early noninvasive approach is taken, clopidogrel is recommended for at least 1 month (IA) and for up to 1 year if possible (IB). If no intervention is performed and the patient has recurrent symptoms, heart failure, or rhythm disturbances, clopidogrel load and daily maintenance dose can be added to current therapy (IA).27
Ticlopidine
Ticlopidine is given at 250 mg twice daily with platelet inhibition starting within 24 to 48 hours. Ticlopidine is an effective antiplatelet agent in ACS patients and clearly demonstrated a reduction compared with placebo in nonfatal MI and combined end point of vascular death and nonfatal MI.41 The reduction in nonfatal MI and death was confirmed in a follow-up substudy in patients with angina at rest and with electrocardiographic changes who received the same dose of ticlopidine for 6 months.42
Ticlopidine is prescribed less frequently than clopidogrel because of side effects, including nausea, vomiting, and diarrhea, but the most serious side effects are hematologic. Neutropenia was reported in 2.4% of patients with severe neutropenia in 0.8%. Even more concerning are the rare reported incidences of thrombotic thrombocytopenia purpura (TTP) associated with ticlopidine use.43
The superior safety profile of clopidogrel over ticlopidine was demonstrated in three comparative studies between ticlopidine and clopidogrel in the post-PCI population. The reduced bleeding complications were achieved with no compromise in efficacy of reducing ischemic complications. In the larger randomized trial, 1020 patients were randomized after coronary stenting to a 28-day regimen of either (1) 300-mg clopidogrel loading dose followed by 75 mg daily; (2) 75 mg daily of clopidogrel; or (3) 250 mg twice daily ticlopidine. All patients received 325 mg of aspirin daily. The primary end point included any of the following: major peripheral or bleeding complications, neutropenia, thrombocytopenia, or early discontinuation of study drug. This occurred in 9.1% of the ticlopidine group compared with 4.6% in the combined clopidogrel group (relative risk 0.50; 95% CI 0.31 to 0.81; p = 0.005). Ischemic complications (cardiac death, MI, and target lesion revascularization were low and comparable between treatment groups (0.9% with ticlopidine, 1.5% with 75 mg/day clopidogrel, 1.2% with the clopidogrel loading dose; p = NS for all comparisons).44
These results were supported in two separate studies. In the first, clopidogrel 75 mg daily or ticlopidine 500 mg daily was initiated just after stent placement in 700 patients. All patients were taking aspirin 100 mg. Therapy was continued for 4 weeks. A primary composite end point of ischemic events within 30 days of stent placement (cardiac death, urgent target vessel revascularization, angiographically documented thrombotic stent occlusion or nonfatal MI) was rare and not significantly different between both groups. Primary noncardiac end points defined as noncardiac death, stroke, severe peripheral vascular or hemorrhagic events, or any events causing discontinuation of the study drug occurred in 16 patients taking clopidogrel and 33 patients taking ticlopidine (p = 0.01).45 Further confirmation that clopidogrel was as effective as ticlopidine in patients receiving intracoronary stents occurred when 30-day event rates were measured in 500 patients receiving coronary stents that were treated with aspirin and clopidogrel (300 mg loading then 75 mg/day for 14 days) to 827 patients with coronary stents treated with aspirin and ticlopidine (500 mg loading dose and 250 mg twice daily for 14 days). Mortality was 0.4% in clopidogrel patients versus 1.1% in ticlopidine patients; nonfatal MI occurred in 0% versus 0.5%, stent thrombosis in 0.2% versus 0.7%, bypass surgery or repeat angioplasty in 0.4% versus 0.5%, and any event occurred in 0.8% versus 1.6% of patients, respectively (p = NS).46
Prasugrel
The newest of the ADP receptor antagonists, prasugrel has recently been approved by the FDA for use in the US. Phase 3 clinical trials suggest it is a more potent platelet inhibitor that may add a new weapon for the treatment of ACS. The loading dose of prasugrel (60 mg) appears more potent than the commonly used 600 mg of clopidogrel. Similarly, the maintenance dose of prasugrel (10 mg) maintains a higher degree of platelet inhibition than a daily dose of 150 mg of clopidogrel.47
The largest clinical investigation to date to examine prasugrel was the TIMI-38 or the TRITON trial.48 In that study, 13,608 patients with moderate to high risk ACS scheduled for PCI were randomized to receive prasugrel (a 60-mg loading dose and a 10-mg daily maintenance dose) or clopidogrel (a 300-mg loading dose and a 75-mg daily maintenance dose) for 6 to 15 months. All patients received aspirin in addition to thienopyridine therapy. The primary efficacy end point was a composite of cardiovascular death, nonfatal MI, or nonfatal stroke, whereas the key safety end point was major bleeding. There was a statistically significant 19% reduction in the primary end point in the prasugrel group (9.9% versus 12.1%, hazard ratio for prasugrel versus clopidogrel, 0.81; 95% confidence interval [CI], 0.73 to 0.90; p <0.001). The use of prasugrel was also associated with significant reduction in other ischemic complications, such as MI and urgent target-vessel revascularization. Interestingly, there was a significant reduction in stent thrombosis as well (1.1% versus 2.4%, p <0.001).48 However, there was a significant concern about the bleeding complications encountered in the TIMI-38 trial. Among patients receiving prasugrel and aspirin, there was a significant increase in the rate of major bleeding complications compared with those receiving clopidogrel (2.4% versus1.8%, hazard ratio 1.32; 95% CI, 1.03 to 1.68, p = 0.03). Even more concerning, a similar statistically significant increase was noted in fatal bleeding (0.4% versus 0.1%; p = 0.002).48 These findings represent a dilemma for prescribing physicians seeking a balance between improved efficacy, but without an increase in the risk of serious complications.
Glycoprotein IIb/IIIa Receptor Antagonists
Platelet aggregation depends on the glycoprotein IIb/IIIa receptor located on the surface of platelets. Often called the final common pathway, the IIb/IIIa receptor on the activated platelet undergoes a conformational change that allows binding of fibrinogen and vWF, which in turn cross-links with IIb/IIIa receptors on other activated platelets promoting platelet aggregation.
Abciximab
Abciximab was the prototype of all intravenous IIb/IIIa receptor antagonists. It is a chimeric human-murine monoclonal antibody that irreversibly binds to the IIb/IIIa receptor. Abciximab also cross reacts with the avß3 (vitronectin) receptor.49 It has a plasma half-life of approximately 30 minutes and is cleared through the reticulo-endothelial system, but once bound to the platelet it remains nearly irreversibly bound and maintains some IIb/IIIa blockade for up to10 to 14 days. The dose of abciximab approved for use in conjunction with other antithrombotics in the setting of PCI is a bolus of 0.25 mg/kg 10 to 60 minutes before intervention, and then given as an infusion of 0.125 μg/kg/min (10 μg/min maximum) for 12 hours.
Numerous landmark trials have established the value of abciximab as an adjunct to heparin for anticoagulation during PCI. Those trials included both stable and unstable patients.50–53 In those trials, the benefit of abciximab in reducing major adverse ischemic end points at 30 days (primarily periprocedural MI and urgent revascularization) was achieved with the use of the bolus and a 12-hour infusion regimen. However, in the earlier randomized trial, CAPTURE, abciximab was used in a different regimen, which resulted in some interesting observations.54 In this trial, 1266 patients with high risk ACS (refractory UA and/or NSTEMI) were randomized to a placebo or abciximab given for 18 to 24 hours before intervention and continuing for 1 hour after the procedure. The primary end point (death, MI, or urgent intervention within 30 days of enrollment) was significantly reduced in the abciximab group (11.3% versus 15.9%, p = 0.012). The observed difference was mainly because of the reduction in MI (4.1% versus 8.2% in favor of the abciximab group, p = 0.002). Interestingly, the benefit of intense antiplatelet therapy was observed within 24 hours, even before revascularization was attempted (Fig. 37-4). Further analysis of the CAPTURE study demonstrated the benefit of intense and early platelet inhibition by abciximab was mostly observed in patients having elevated serum troponin levels (see Fig. 37-3).55
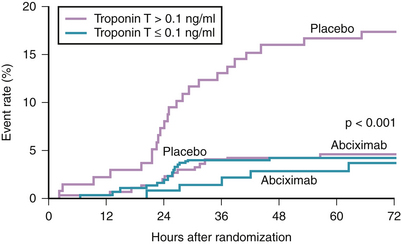
In contrast to the aforementioned studies, the GUSTO IV-ACS study evaluated the efficacy of abciximab therapy in patients with chest pain who were troponin positive or had 0.5 mm of ST depression and were not undergoing early revascularization. Somewhat surprisingly, there was no reduction in death or MI at 30 days in patients with ACS receiving abciximab as a bolus plus 24 to 48 hour infusion after presentation. In fact, at 48 hours, a higher rate of mortality was seen in patients receiving abciximab.56 This observation (excess mortality with abciximab therapy) remained true after 1 year follow-up in patients having a low troponin or elevated c-reactive protein.57 The reason for this excess mortality is unclear. One possibility is that subtherapeutic dosing or insufficient platelet inhibition by IIb/IIIa inhibitors may actually be harmful in patients with higher levels of inflammation.58 The antiplatelet effects of abciximab become unpredictable and tend to diminish with time. This was found in a small pharmacodynamic study in 100 patients where platelet function was tested using a rapid platelet function assay (VerifyNow) immediately after an abciximab bolus, 8 hours into a 12-hour abciximab infusion, and 13 to 26 hours after the bolus. After the bolus, abciximab achieved 95% platelet inhibition. At 8 hours platelet inhibition had decreased to 88 ± 9% with 13 patients achieving less than 80% inhibition. Between 13 and 26 hours (mean 19), platelet inhibition had dropped to a mean of 71 ± 14%.59 This failure to sustain an adequate degree of platelet inhibition may explain the excess mortality in patients receiving 24 to 48 hours of abciximab infusion in the GUSTO IV-ACS study.
In the current era of routine use of thienopyridines and aspirin to treat ACS patients, the question has been raised as to the role of IIb/IIIa antagonists in patients already receiving dual antiplatelet therapy. This question was examined in two clinical trials addressing the role of abciximab with PCI in low- and high-risk patients: ISAR REACT and ISAR REACT II. In ISAR REACT, 2159 patients undergoing elective PCI were randomized to receive abciximab or placebo. All patients received clopidogrel 600 mg 2 hours before the procedure and the composite end point was death, MI, or target vessel revascularization within 30 days. The primary end point was achieved in 4% of patients in both groups, suggesting that abciximab provided no additional benefit in low- to intermediate-risk patients undergoing elective PCI who are pretreated with clopidogrel.60 Maximum antiplatelet therapy is desired in patients with ACS undergoing PCI because platelet aggregates can cause microemboli, leading to periprocedural MI and sudden cardiac death. Therefore the benefit of abciximab, when added to clopidogrel, may only be demonstrated in patients having an unstable plaque, as opposed to more stable atherosclerotic disease as seen in the patients studied in ISAR REACT. This hypothesis was tested in the ISAR REACT II trial, which randomized 2022 patients having acute coronary syndromes to receive abciximab or a placebo—after all patients were pretreated with heparin, aspirin, and 600 mg of clopidogrel. Again, the composite end point was death, myocardial infarction, or target vessel revascularization. The target end point was reached in 8.9% of the abciximab group and 11.9% of the placebo group (relative risk 0.75, 95% CI, 0.58 to 0.97; p = 0.03). The patients who benefited from abciximab had an elevated troponin, suggesting that abciximab is beneficial only in high-risk patients who already demonstrate evidence of active atherothrombotic disease with distal embolization.
Eptifibatide
The benefit of eptifibatide in patients with acute coronary syndrome was established in the PURSUIT trial. This randomized placebo-controlled trial included 10,948 ACS patients without STEMI. Most patients received aspirin and heparin, and were then randomized to receive a placebo or eptifibatide (intravenous bolus of 180 μg/kg followed by an infusion at 1.3 or 2.0 μg/kg/min). The duration of therapy extended to 72 hours (or 96 if intervention occurred later in the hospitalization) or until the patient left the hospital. The primary end point was a composite of death and nonfatal MI at 30 days. In the eptifibatide group, there was a 1.5% absolute reduction in the incidence of the primary end point (14.2% versus 15.7%, p = 0.04), with this effect occurring in most major subgroups.43,61 Further investigation of the PURSUIT cohort reveals that eptifibatide reduced the rates of death and MI in patients before and after PCI, in stented and nonstented patients and those who did not undergo PCI, though the reduction in primary events appears greater in patients who had an early PCI.62
Tirofiban
Related posts:
 Evolution of the Coronary Care Unit: Past, Present, and Future
Evolution of the Coronary Care Unit: Past, Present, and Future
 Elevated Cardiac Troponin in the Absence of Acute Coronary Syndromes: Mechanism, Significance, and Prognosis
Elevated Cardiac Troponin in the Absence of Acute Coronary Syndromes: Mechanism, Significance, and Prognosis
 Conduction Disturbances in Acute Myocardial Infarction
Conduction Disturbances in Acute Myocardial Infarction
 Pharmacologic Interactions in the CICU
Pharmacologic Interactions in the CICU
 Acute Aortic Syndromes: Diagnosis and Management
Acute Aortic Syndromes: Diagnosis and Management
 Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


