CHAPTER 36 Anticoagulation
Antithrombin Therapy
Population-based surveys conducted in Europe during 1999-2001 demonstrated that more than 90% of patients with ACS receive aspirin upon hospital admission and that most (80%) also receive either UFH or LMWHs, with the proportions receiving these two forms of heparin approximately equal.1–3
The Seventh American College of Chest Physicians (ACCP) Consensus Conference on Antithrombotic Therapy and the American College of Cardiology/American Heart Association/ Society for Cardiovascular Angiography and Interventions (ACC/AHA/SCAI) updated their recommendations for anticoagulation therapy in patients undergoing PCI in recent years and the European Society of Cardiology has also recently published its position paper on anticoagulants in heart disease.4–6
Hemostasis and the Coagulation Cascade
When a blood vessel is damaged, the site of disruption is rapidly sealed to prevent blood loss. Hemostasis refers to the formation of a platelet and fibrin plug at the site of injury. This requires activation of platelets and coagulation at the site of injury. The clot is later dissolved by another protease reaction, fibrinolysis, which also prevents the vessel from being occluded by the clot during its formation. In order for the blood to stay fluid within the circulation, a delicate balance between the carefully regulated systems of coagulation and fibrinolysis is needed. Disturbances in either system will cause a tendency toward thrombosis or bleeding.7 Coagulation, as shown in Figure 36-1, has often been represented as two independent pathways that converge to a common pathway, with thrombin generation as the end point of the cascade. This model gives a good representation of the processes observed in clinical coagulation laboratory tests. The prothrombin time measures the factors of the so-called extrinsic pathway, and activated partial thromboplastin time measures factors in the intrinsic pathway. However, this model has inadequacies relating to the in vivo hemostatic processes. For example, deficiencies of factor XII, high molecular weight kininogen (HK), or prekallikrein do not cause clinical bleeding, and others have shown that under normal circumstances, hemostasis is initiated by tissue factor (TF), a transmembrane glycoprotein.8,9 TF is a member of the class II cytokine receptor superfamily and functions both as the receptor and as the essential cofactor for factors (F) VII and VIIa. Assembly of the TF/FVIIa complex on cellular surfaces leads to the activation of FX and initiates coagulation. TF is constitutively expressed in cells surrounding blood vessels and large organs to form a hemostatic barrier. In addition to its role in hemostasis, the TF/FVIIa complex has been shown to elicit intracellular signaling resulting in the induction of various genes, thus explaining its role in various biologic functions, such as embryonic development, cell migration, inflammation, apoptosis, and angiogenesis.10–12
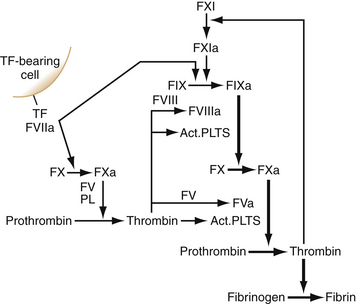
Cell-based Model of Coagulation
In this model, coagulation occurs in three overlapping phases: initiation, priming, and propagation.9,13,14 During the process of hemostasis, a break in the vessel wall brings plasma into contact with TF-bearing cells. FVII binds to TF and is rapidly activated by coagulation proteases and by noncoagulation proteases, depending on the cellular location of the TF.15–17 The FVIIa/TF complex then activates FX and FIX. The activated forms of these two proteins (IXa and Xa) play very different roles in subsequent coagulation reactions. FXa can activate plasma FV on the TF cell. If FXa diffuses from the protected environment of the cell surface from which it was activated, it can be rapidly inhibited by the TF pathway inhibitor (TFPI) or antithrombin (AT). However, the FXa that remains on the TF cell surface can combine with FVa to produce small amounts of thrombin (the enzyme responsible for clot formation). This thrombin, although not sufficient to cleave fibrinogen throughout a wound, nonetheless plays a critical role in amplifying the initial thrombin signal. The initial FVIIa/TF complex is subsequently inhibited by the action of the TFPI in complex with FXa.18,19 In contrast to FXa, FIXa is not inhibited by TFPI, and only slowly inhibited by AT. FIXa moves in the fluid phase from TF-bearing cells to nearby platelets at the injury site.
In the amplification phase (priming phase), low concentrations of thrombin activate platelets adhering to the injury site to release FV from their α-granules. Thrombin cleaves the partially activated FV to a fully active form. Thrombin also cleaves FVIII, releasing it from the von Willebrand factor. Such activated factors bind to platelet surfaces, which provide the backbone for thrombin generation that occurs during the propagation phase.20
Arterial Thrombosis
Arterial thrombosis can occur from at least two main different mechanisms, endothelial erosion or plaque rupture.21,22 Plaque rupture results in the exposure of thrombogenic material (e.g., collagen and TF) to the circulation and subsequent activation of platelets and coagulation cascade. This, coupled with the simultaneous release of vasoactive substances, induces thrombus formation and vasoconstriction, which result in myocardial ischemia and ACS.
Antithrombins—Mechanism of Action
Thrombin has an active site and two exosites, one of which, exosite 1, binds to its fibrin substrate, orienting it toward the active site. Figure 36-2 displays the mechanisms of action of the different thrombin inhibitors described below. The heparin derivatives in current use include UFH, LMWHs, and the synthetic pentasaccharide derivatives fondaparinux and idraparinux. These are all parenteral drugs that must be administered by intravenous (IV) or subcutaneous (SC) injection, and they are classified as indirect anticoagulants because they require a plasma cofactor (essentially AT) to exert their anticoagulant activity, while the DTIs (hirulog and hirudin) bind thrombin, essentially displacing it from fibrin.
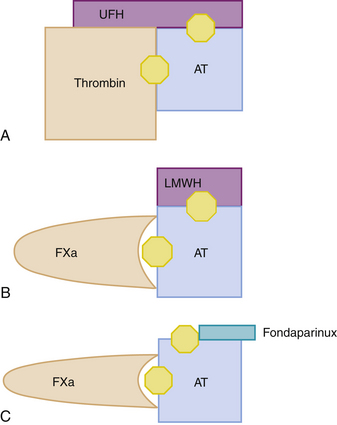
Unfractionated Heparin
UFH was discovered almost 90 years ago and is the prototype of its derivatives. It is a natural product that can be isolated from beef lung or porcine intestinal mucosa. It consists of highly sulfated polysaccharide chains, with a mean of about 45 saccharide units. Only one third of the heparin chains possess a unique pentasaccharide sequence that exhibits high affinity for antithrombin (AT), and it is this fraction that is responsible for most of the anticoagulant activity of heparin.5
Pharmacokinetics, Metabolism, and Administration
UFH must be given parenterally either by continuous IV infusion or by SC injection. When given SC for treatment of thrombosis, higher doses of heparin than those administered by IV infusion are needed to overcome the fact that the bioavailability of heparin after SC injection is only about 30%.21 This is, however, highly variable among individuals.5 A number of plasma proteins compete with AT for heparin binding, thereby reducing its anticoagulant activity. The levels of these proteins vary among patients. This phenomenon contributes to the variable anticoagulant response to heparin and to the phenomenon of heparin resistance.21 Heparin is cleared through a combination of a rapid saturable phase and a slower first-order mechanism. Heparin binds to endothelial cells, platelets, and macrophage during the saturable phase. Once the cellular binding sites are saturated, heparin enters the circulation, from where it is cleared more slowly via the kidneys. The complex kinetics of heparin clearance render the anticoagulant response to UFH nonlinear at therapeutic doses, with both the peak activity and duration of effect increasing disproportionately with increasing doses.21
Dosing and Monitoring
Heparin can be given in fixed or weight-adjusted doses, and nomograms have been developed to aid with dosing.22 The doses of UFH recommended for the treatment of ACS are lower than those typically used to treat venous thromboembolism due to the lower thrombus burden in arterial thromboses. Due to the varying anticoagulant response of UFH among patients, UFH therapy is monitored and the dose is adjusted based on these results. The test most often used to monitor heparin is the activated partial thromboplastin time (aPTT). The activated clotting time (ACT) is used to monitor the higher doses of UFH given to patients undergoing PCI or cardiopulmonary bypass surgery.
An aPTT ratio between 1.5 and 2.5 (calculated by dividing the reported therapeutic aPTT range by the control value for the reagent) was associated with a reduced risk for recurrent VTE in a previous large retrospective registry.23 Based on this study, an aPTT ratio of 1.5 to 2.5 was adopted as the therapeutic range for UFH. Both the ACCP and the European Society of Cardiology agree that aPTT ratios should be adapted to the reagents used and that the use of a fixed aPTT target in seconds for any therapeutic indications of UFH should not be strictly applied. Despite these shortcomings, the aPTT is the most common method used for monitoring its anticoagulant response. The aPTT should be measured approximately 6 hours after the bolus dose of heparin, and the continuous IV dose should be adjusted according to the result. Various heparin dose-adjustment nomograms have been developed, but none is applicable to all aPTT reagents, and, for the reasons discussed above, the therapeutic range should be adapted to the responsiveness of the reagent used.
Side Effects
Bleeding is the major complication of heparin therapy. Other complications of heparin include heparin-induced thrombocytopenia (HIT) and osteoporosis. HIT is caused following the formation of heparin/platelet factor 4 (PF4) complexes. Antibodies are produced against the neoepitope on PF4 that binds to Fc receptors on the platelet, causing its activation.24 Activated platelets are then removed from the circulation, which causes thrombocytopenia. Osteoporosis is a complication of long-term treatment with therapeutic doses of heparin and appears to be the result of heparin binding to osteoblasts with subsequent osteoclast activation.
Clinical Evidence
UFH has mainly been used in the management of patients with non–ST-segment elevation-ACS (NSTE-ACS), adjuvant therapy during PCI, and in the STEMI population (with or without fibrinolytic agents). In trials comparing the association of heparin plus aspirin versus aspirin alone in NSTE-ACS, a trend toward benefit was observed in favor of the heparin-aspirin combination, but at the cost of an increase in bleeding. Recurrence of events after interruption of UFH explains why this benefit is not maintained over time, unless the patient was revascularized before the interruption of UFH.25 Adjunctive UFH use in the setting of STEMI, together with the use of fibrinolytic agents, has a narrow therapeutic window. The ISIS-3 and GISSI-2 trials examined its use with streptokinase and found no reduction in mortality at 35 days or 6 months with an increase in major bleeding. The evidence for the use of UFH with tissue plasminogen activator (tPA) was more favorable and it became standard therapy after the superiority of front-loaded tPA with UFH was demonstrated in the GUSTO-1 trial.26 Intravenous UFH using a bolus of 60 U/kg (maximum 4000 U) followed by a maintenance infusion of 12 U/hr (maximum 1000 U) for 48 hours is recommended with tPA and other fibrin-specific fibrinolytic agents.5
UFH is the most commonly used anticoagulant during PCI. ACT monitoring is used in this case because the required level of anticoagulation is beyond the range that can be measured using aPTT. Randomized trials have shown UFH to reduce ischemic complications.27,28 UFH in a dose of 60 to 100 IU/kg and a target ACT between 250 and 350 seconds are recommended. A target of 200 seconds is advocated for UFH dosing in conjunction with glycoprotein (Gp) IIb/IIIa inhibitors.29–32 After completion of PCI, UFH is not indicated because continued treatment does not reduce ischemic complications and is associated with a higher risk of bleeding.33
Low Molecular Weight Heparin
In the early 1980s, LMWH was introduced as an alternative to UFH, first for the prevention of deep vein thrombosis (DVT) among postoperative patients.34,35 Four LMWHs have been studied in the setting of acute coronary syndromes, enoxaparin, reviparin, tinzaparin, and dalteparin. Enoxaparin and dalteparin have been approved by the FDA for use in cardiovascular disease, and enoxaparin is the most widely studied of the LMWHs.
The high bioavailability and minimal plasma protein binding of LMWH leads to a predictable dose-response, which obviates the need for plasma monitoring.36 In addition, LMWH has been reported to have a lesser effect on platelet aggregation than UFH.37 Other benefits of LMWH include a higher antifactor Xa:IIa ratio, less inhibition by platelet factor 4, and an inhibition in the early rise in von Willebrand factor.38
Pharmacokinetics, Metabolism, and Administration
The LMWHs are most commonly administered SC, although an IV bolus has also been evaluated, most commonly in studies in the STEMI population. Enoxaparin is dosed according to body weight, with the most common dose used in ACS being 1 mg/kg every 12 hours. Following a single SC injection, enoxaparin has a peak plasma antifactor Xa activity within 3 to 6 hours.39 One study demonstrated that 97.6% of patients had antifactor Xa activity above therapeutic range at the time of catheterization if enoxaparin was administered with 8 hours of planned PCI.40 Enoxaparin is weakly metabolized in the liver. Renal clearance of active fragments represents about 10% of the administered dose, whereas about 40% of active and inactive fragments combined are excreted renally. Based on antifactor Xa activity, elimination half-life is 4.5 to 5 hours after a single dose and approximately 7 hours after repeated dosing.41
Clinical Evidence
Based on early experience with LMWHs,42–44 two pivotal trials examined the efficacy and safety of enoxaparin in patients with ACS. The ESSENCE trial evaluated enoxaparin in the setting of ACS.45 They randomized more than 3000 patients to enoxaparin or UFH for at least 48 hours. At 14 days, the incidence of death, MI, or recurrent angina was significantly lower in patients randomized to enoxaparin than those receiving UFH (16.6% versus 19.8%, p = 0.019), and the outcomes remained significantly different at 30 days (19.8% versus 23.3%, p = 0.016). Although there was no significant difference in major bleeding (6.5% versus 7.0%, p = NS), the incidence of any bleeding event was significantly higher in the enoxaparin group (18.4% versus 14.2%, p = 0.001), primarily driven by an increase in injection-site ecchymosis. The TIMI 11B trial, which randomized nearly 4000 patients to either UFH or enoxaparin, again showed a statistically significant difference in ischemic outcomes favoring enoxaparin (12.4% versus 14.5%, p = 0.048) with no difference in major bleeding.46
The SYNERGY trial randomized more than 10,000 patients having NSTE-ACS and high risk features undergoing an early invasive strategy to enoxaparin versus UFH.47 More than half received a Gp IIb/IIIa inhibitor and two thirds received clopidogrel or ticlopidine. There was no significant difference in the incidence of death or nonfatal MI by 30 days between patients who received enoxaparin or UFH (14.0% versus 14.5%, p = NS). In addition, the rates of acute complications, including abrupt closure, threatened abrupt closure, unsuccessful PCI, and emergency CABG surgery were not significantly different between the groups. There was, however, a significantly higher incidence of major bleeding (9.1% versus 7.6%, p = 0.008) in the enoxaparin group.
There have also been two large meta-analyses reported evaluating the role of enoxaparin in NSTE-ACS.48,49 Eikelboom and colleagues analyzed five trials involving more than 12,000 patients comparing UFH with LMWH. They reported no significant difference between LMWH and UFH in terms of 7-day incidence of a composite of death or recurrent MI (2.2% versus 2.3%, p = 0.34).48 Petersen and colleagues evaluated six studies, including almost 22,000 patients randomized to either enoxaparin or UFH, and reported their results in 2004.49
Related posts:
 Evolution of the Coronary Care Unit: Past, Present, and Future
Evolution of the Coronary Care Unit: Past, Present, and Future
 Conduction Disturbances in Acute Myocardial Infarction
Conduction Disturbances in Acute Myocardial Infarction
 Diagnosis and Treatment of Complications of Coronary and Valvular Interventions
Diagnosis and Treatment of Complications of Coronary and Valvular Interventions
 Pharmacologic Interactions in the CICU
Pharmacologic Interactions in the CICU
 Acute Aortic Syndromes: Diagnosis and Management
Acute Aortic Syndromes: Diagnosis and Management
 Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


