CHAPTER 40 Antiarrhythmic Electrophysiology and Pharmacotherapy
Despite the important role that antiarrhythmics play to combat cardiac arrhythmias, it has been difficult to overcome the stigma associated with these drugs. This stigma has stemmed from earlier published trials such as the CAST and CAST II trials evaluating class IC antiarrhythmics and the SWORD trial evaluating sotalol in patients with coronary artery disease and left ventricular dysfunction. These trials demonstrated increased mortality, significant side effects, and marginal efficacy.1,2,3 The Achilles heel of these agents is their propensity for proarrhythmia that results in more dangerous arrhythmias than the ones they were initially intended to treat. This led to a re-evaluation of the wisdom of using relatively simple agents that took aim at single molecular targets, such as sodium or potassium channels. Subsequently, researchers pursued the development of newer antiarrhythmic agents that target multiple molecules or complex arrhythmogenic pathways, such as amiodarone or d,l-sotalol. There was a renewed interest and emphasis on the use of β-blockers, which do not specifically target ion channels. In addition there has been significant interest in understanding the genetic basis of susceptibility to serious arrhythmias in individuals with genetic syndromes such as long QT and Brugada syndromes.
His-Purkinje Action Potential
The His-Purkinje action potential can be conceptually divided into periods of depolarization, repolarization, and resting states. However, the action potential is traditionally divided into five different phases (phases 0-4) to describe the activity of various ion channels that bring about any of these three states (Fig. 40-1, A and B). Phase 4 corresponds to the resting state when the cell is not being stimulated and is ready for subsequent depolarization. Phase 0 corresponds to depolarization of the myocardial cell. It initiates a cascade of events involving the influx and efflux of multiple ions, leading to phases 1-3, manifesting in repolarization and refractoriness.
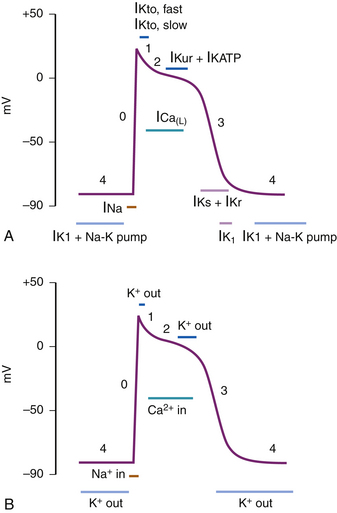
Phase 4
The normal resting membrane potential in the ventricular myocardium is between −85 to −95 mV. This membrane potential is determined by the balance of inward sodium (Na+) and calcium (Ca2+) currents and outward potassium (K+) current. The equilibrium potential for a given ion is determined by the concentrations of that ion inside and outside the cell. K+ is the principal cation intracellularly, whereas phosphate and conjugate bases of organic acids are the dominant anions. Extracellularly, Na+ and chloride (Cl−) are the principal cation and anion, respectively. Therefore, when K+ channels open, K+ ions flow outside the cell along their concentration gradient, leaving the cell with a more negative membrane potential.
This phase of the action potential is associated with cardiac diastole.
Phase 2 (Plateau)
The plateau phase is unique to the cardiac cell and represents an interruption to rapid repolarization, extending the duration of the action potential and therefore the refractoriness of the cardiac cell. The benefit of this is to allow for a single contraction of myocardium to occur before the generation of a subsequent action potential, thus the heart can never be “tetanized,” which would be incompatible with cardiac function.4
SA Node and AV Node Action Potential
SA and AV nodal action potentials are very similar with only minor differences between them in phase 0. They are significantly different from fast response tissue’s action potentials previously described. These slow response tissue action potentials are divided into three phases instead of five (Fig. 40-2, A and B). Phase 4 is the spontaneous depolarization (pacemaker potential) phase that triggers the action potential once the membrane potential reaches threshold (between −40 and −30 mV). Phase 0 is the depolarization phase of the action potential. This is followed by phase 3 repolarization. Once the cell is completely repolarized at about −60 mV, the cycle is spontaneously repeated.
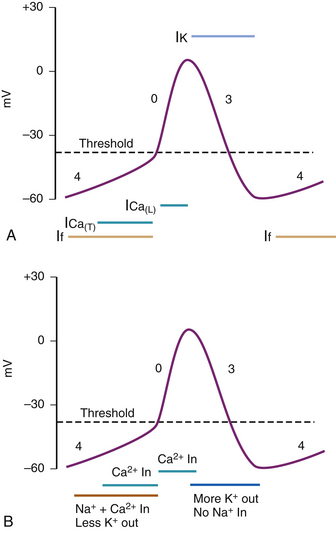
Phase 0—Depolarization
The upstroke (Vmax) and therefore the conduction velocity in a pacemaker cell is significantly slower than in the fast response tissue cells, with the AV node action potential demonstrating a faster Vmax than that of the SA node.5
Classification
Vaughan-Williams
This is currently the most widely used classification of antiarrhythmic drugs (Table 40-1). This classification was initially introduced by Vaughan Williams6,7 and was based on the electrophysiologic effects of antiarrhythmics. It was later modified by Harrison8 with the recognition that drugs in the same class have different potencies and that the same drug may exert multiple class effects.
Table 40–1 Vaughan-Williams Classification of Antiarrhythmics and Their Mechanism of Action
| Class | Mechanism | Drugs |
|---|---|---|
| I | Na+ channel blockers | |
| A | Slows conduction velocity (Vmax) and prolongs action potential. Results in decreased conductivity and increased refractoriness. | Quinidine, procainamide, disopyramide |
| B | Minimal effect on conduction velocity (Vmax) and shortens action potential duration. Results in decreased refractoriness. | Lidocaine, mexiletine |
| C | Significant slowing of conduction velocity (Vmax), minimal effect on action potential duration. Results in decreased conductivity and no change in refractoriness. | Flecainide, propafenone, moricizine |
| II | Atenolol, esmolol, propranolol | |
| III | Amiodarone, sotalol, dofetilide, ibutilide, azimilide | |
| IV | Verapamil, diltiazem (non-dihydropyridine) | |
| V | Digoxin, adenosine |
Amiodarone has effects across all classes, blocking Na+ channels in depolarized tissues, but is also capable of affecting Ca2+ and K+ channels and adrenergic receptors. This makes it an extremely versatile drug capable of affecting a wide variety of arrhythmias. Sotalol has a significant β-blocker effect in addition to its class III action. Ibutilide can also enhance the slow delayed sodium current. Although moricizine has diverse effects across different classes, this drug is not currently used because it has been demonstrated to increase mortality.
Class I Antiarrhythmics and Use Dependence
Class I drugs are all Na+ channel blockers with varying potency and are classified based on their effect on the action potential upstroke or Vmax and therefore their ability to alter conduction velocity. They can prolong it, shorten it, or have no net effect (Fig. 40-3). Their different potency is due to variable rates of binding and dissociation from the channel receptor.9 Class IC are the most potent Na+ channel blockers because they have the slowest binding and dissociation from the receptor; class IA are the least potent (fastest binding and dissociation); and class IB are moderately potent. Faster heart rates allow less time for drug-receptor dissociation, resulting in an increased total number of blocked receptors and more effective antiarrhythmic action. This is known as “use dependence” and is most noted with class IC agents.10 These effects can result in prolonged conduction velocity and manifest by widening of the QRS complex on the surface electrocardiogram (ECG), especially during tachycardia.
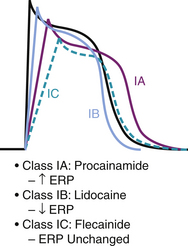
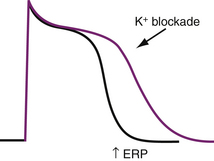
Class III Antiarrhythmics and Reverse Use Dependence
Class III antiarrhythmic agents extend the plateau phase of the action potential by blocking K+ channels. They effectively prolong repolarization and the action potential duration with a resultant increase in refractoriness without a change in conductivity (Fig. 40-4). This manifests as prolongation of the Q–T interval on the surface ECG. These effects are most pronounced during slow heart rates. This is known as “reverse use dependence” and thus, longer Q–T intervals are noted at slower heart rates.11 This provides the potential for dangerous arrhythmias such as torsades de pointes.12 This is known as proarrhythmia and is one of the most serious, life-threatening side effects associated with antiarrhythmic drug use. Amiodarone, however, appears to be the exception, with proarrhythmia being reported only uncommonly.13
Class II Antiarrhythmics
These drugs act by competitive inhibition of the β-adrenergic receptor, and largely affect the SA and AV node. Yet they also have mild Na+ channel inhibitory effects. There is a preponderance of data demonstrating the antiarrhythmic properties of β-blockers. β-blockers have played an increasingly important role in increasing survival in patients with coronary heart disease and congestive heart failure, along with adjunctive therapy to implantable cardiac defibrillators (ICDs) by decreasing the incidence of sudden cardiac death (SCD). Although not all of the beneficial effects of β-blockers are understood it is likely that they essentially work by reversing or preventing the proarrhythmic actions of sympathetic activity.14 These include increased automaticity because of enhanced phase 4 depolarization in the SA and AV nodes, increased membrane excitability in phase 2 and 3 of the His-Purkinje action potential, increased Vmax, and increased delayed afterpotentials, which can lead to increased triggered activity type arrhythmias. They are therefore most effective in tissue under intense adrenergic stimulation (e.g., in ischemia) and it is not surprising that their effects are most obvious in patients having acute myocardial infarction and decompensated heart failure.
Class IV
The non-dihydropyridine calcium channel blockers (verapamil and diltiazem) are the only ones that exhibit significant electrophysiologic and antiarrhythmic properties. These are mediated through their ability to block the slow calcium channel. Their antiarrhythmic properties are exerted through two main effects. Their most prominent activity is to block the slow calcium channel in the action potential of the SA and AV node, effectively slowing phase 4 spontaneous depolarization. This results in a variable slowing of the heart rate (similar to β-blocker–induced adrenergic blockade)15 and slowing conduction through the AV node, manifested by bradycardia and prolongation of the P-R interval on the surface ECG.
The other action is their ability to shorten the plateau phase of the action potential of ventricular myocytes, therefore inhibiting early afterdepolarizations (EADs).16 EADs are due to fluxes in calcium and appear to occur mostly under conditions that prolong the action potential (e.g., hypoxia, drug induced), giving rise to torsades de pointes. Therefore, calcium channel blockers may play an active role in the prevention of these types of arrhythmias.
Sicilian Gambit
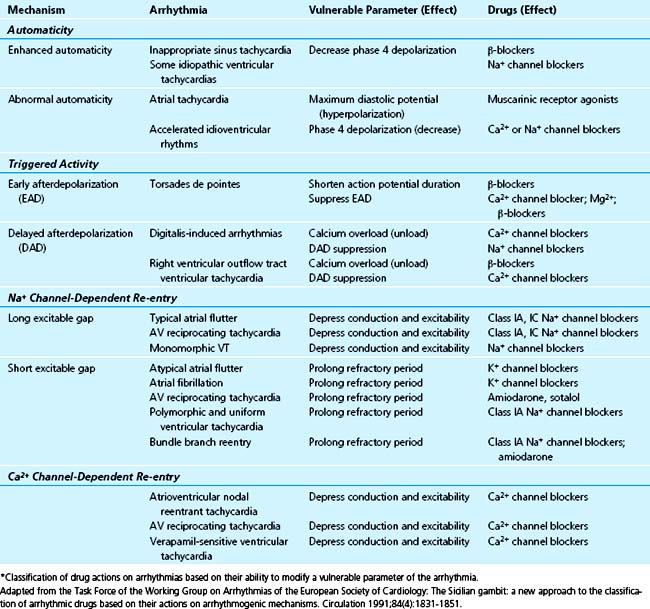
Although the merits of the Vaughan-Williams classification are its simplicity and wide recognition, providing a useful mode for communication regarding the use of antiarrhythmics, it does not take into account several complexities. These include drugs’ ability to exert effects that cross into different classes, having variable potencies within the same class, and causing other effects such as changes in metabolism, autonomic stimulation, or hemodynamics. The Sicilian gambit17 (Table 40-2) was introduced as a method of rationalizing the approach to the use of antiarrhythmics. Although not widely known and far more complex in its original format, it is included here for its emphasis on approaching antiarrhythmic drug choice based on the mechanism of each individual arrhythmia and the perceived “vulnerable parameter” that can be affected to terminate it.
Pharmacology
Drug Absorption
With rare exceptions, most drugs administered in the critical care setting are administered in the intravenous form and therefore many of the issues surrounding drug absorption do not apply in this circumstance.
Distribution
A classic example is lidocaine administration in heart failure patients. The Vd may be reduced by 40%18 because of decreased perfusion and therefore unless lidocaine bolus doses are reduced in these patients, lidocaine toxicity can result.
Most antiarrhythmics are bound to α1-glycoprotein. This protein’s concentration increases with acute and critical illness (such as shock, trauma, or severe bacterial infection). The result may be decreased effectiveness of the antiarrhythmic agent despite an unchanged plasma concentration, as the fraction of unbound (free) active drug is smaller.
Metabolism and Elimination
Most antiarrhythmics are metabolized hepatically by the cytochrome P450 system, and many undergo extensive first pass metabolism. This accounts for the large ratio of oral to intravenous doses of such medicines as verapamil, metoprolol, or propafenone. Others such as lidocaine are so completely eliminated by the first pass effect that oral dosage is useless.19
A drug’s half-life describes the time required to reduce a plasma drug concentration by 50%. This may be secondary to either drug metabolism and elimination or redistribution. It typically requires five half-lives to eliminate a drug from the plasma completely and five half lives to achieve a steady state. Since only three doses are required to achieve greater than 90% steady state plasma concentrations, drug loading doses should only be used when such an interval is clinically unacceptable such as in life-threatening arrhythmias in critically ill patients. In such situations, a loading dose may be given to achieve desirable plasma concentration levels much faster.20
Antiarrhythmics of Clinical Relevance in the CCU
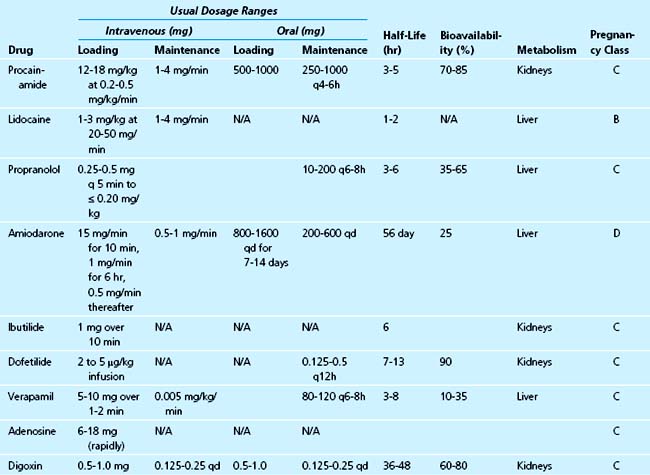
Although detailed accounts of every antiarrhythmic are available elsewhere, the focus of this review is to discuss the antiarrhythmics that have the most clinical relevance to the management of the critically ill patient in the acute care setting. Older medications no longer in current use and those that cannot be administered intravenously or are contraindicated in critical illness are not discussed (Table 40-3).
Class IA
Procainamide
Procainamide is a Na+ channel blocker that results in reduced Vmax associated with decreased conduction velocity and prolongation of the His-Purkinje action potential, and therefore the effective refractory period.21 Its effect on prolongation of the action potential is mediated through its active metabolite N-acetylprocainamide (NAPA), which is a class III antiarrhythmic agent. In addition it can suppress automaticity in slow response tissues (SA and AV nodes)22 and triggered activity in normal Purkinje fibers.23
Related posts:
 Evolution of the Coronary Care Unit: Past, Present, and Future
Evolution of the Coronary Care Unit: Past, Present, and Future
 Elevated Cardiac Troponin in the Absence of Acute Coronary Syndromes: Mechanism, Significance, and Prognosis
Elevated Cardiac Troponin in the Absence of Acute Coronary Syndromes: Mechanism, Significance, and Prognosis
 Conduction Disturbances in Acute Myocardial Infarction
Conduction Disturbances in Acute Myocardial Infarction
 Antiplatelet Therapy
Antiplatelet Therapy
 Acute Aortic Syndromes: Diagnosis and Management
Acute Aortic Syndromes: Diagnosis and Management
 Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


