CHAPTER 41 Analgesics, Tranquilizers, and Sedatives
THE CORONARY care unit (CCU) has evolved drastically over the past several decades from an area primarily limited to the management of the acute coronary insufficiency patient to a full-fledged cardiac intensive care unit (CICU) concerned with a multitude of life-threatening cardiovascular diseases. With this development has also occurred the parallel need for more adequate and flexible analgesia and sedation because many of these critically ill patients undergo invasive procedures, such as balloon angioplasty and placement of intra-aortic balloon pumps, pulmonary artery catheters, and ventricular assist devices. In addition, many require long-term mechanical ventilation. The goal of therapy in the CICU is frequently more complicated than the simple relief of myocardial ischemia or infarction pain and the anxiety that accompanies it. For example, today’s CICU practitioner is more likely to encounter an older and frailer patient, and although the elderly may benefit greatly from analgesic or sedative medications, they are also more likely to suffer the side effects of these medications.1 Likewise, CICU practitioners today are likely to encounter patients who require sedation for long-term mechanical ventilation.
Opioid Analgesics
Morphine
History and Structure
Morphine has been used for analgesia and sedation for many centuries, but it has only been used by the intravenous route for approximately 150 years.4 Morphine is the only naturally occurring opiate agent obtained from the poppy plant, Papaver somniferum, still used with frequency in the critical care setting. Its proper chemical class is the phenanthrene group, which it shares with the agent codeine.5 Morphine occurs in essentially a rigid, pentacyclic type of structure.
Site of Action and Receptor Physiology
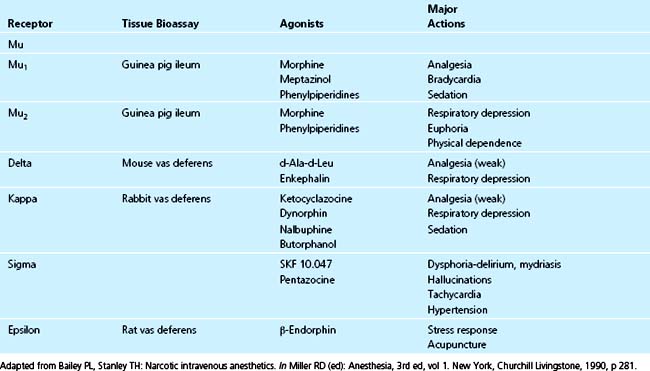
Since the first description of the opiate receptor in 1973, the mechanism for sedation and pain relief has been investigated.6 The initial system included only the μ-receptor; since then, a number of different opiate receptors have been discovered and named, including β-, к-, σ-, and ε-receptors (Table 41-1).7–9 Additional investigations have shown that the μ-receptor system actually includes two subtypes: the μ1-receptor and the μ2-receptor. Although research is ongoing, it appears that the μl-receptor is responsible primarily for analgesia and sedation, whereas the μ2-receptor may be responsible for euphoria and addiction in addition to analgesia.9,10 Both the δ- and к-receptors may be responsible for mediating respiratory depression. The σ-receptor seems to be related to those physiologic responses usually associated with acute withdrawal syndromes, specifically dysphoria, hallucinations, mydriasis, tachycardia, and hypertension.11 Based on these findings, opioid agents have been designed as selective agonists of certain receptors to maximize beneficial physiologic effects while minimizing deleterious ones. The greater μ-specificity of the synthetic opioid fentanyl compared with morphine is an example. Furthermore, agents with partial antagonist properties (e.g., nalbuphine) have been designed to simultaneously act on several opiate receptors, in this case the к- and μ-receptors, to prevent respiratory depression while allowing analgesia.
The different opiate receptors are located in diverse anatomic sites throughout the central nervous system so it is apparent that anatomy and receptor type dictate the pharmacodynamic effects of opiate agents. Whereas opiate receptors are located in a number of distinct areas in the brain and spinal cord, the highest density of receptors is located in the substantia gelatinosa of both the cerebral cortex and spinal cord, the periaqueductal gray areas, the thalamus, and the hippocampus.12 Of these areas, laminae II through V of the substantia gelatinosa and the periaqueductal gray areas appear to have the highest concentrations of μ-receptors and hence the greatest input with respect to analgesia. As a general rule, the gray matter of the central nervous system contains more opiate receptors than does the white matter.13
Pharmacokinetics, Pharmacodynamics, and Metabolism
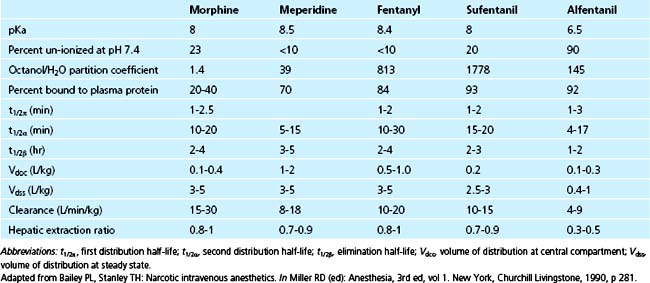
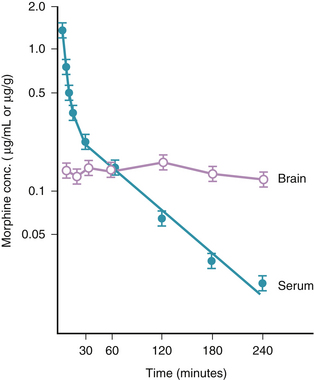
Morphine has a rapid initial redistribution phase of 1 to 1.5 minutes and an initial elimination half-life (t1/2α) of 10 to 20 minutes. Its terminal elimination half-life (t1/2β) is much longer (between 2 and 4.5 hours).14 Morphine is unique compared with the other commonly used opioid agents in that it has relatively low lipid solubility (Table 41-2). Interestingly, the shape of the morphine elimination curve is nearly identical to that of fentanyl, even though it is well known that the clinical behavior of the two drugs (e.g., onset time, offset time, duration of action) is quite different. This is explained by the following comparison between fentanyl and morphine, which is intended to illustrate how two drugs with similar pharmacokinetic properties can have different pharmacodynamic profiles. Compared with fentanyl, morphine has low lipid solubility (it is approximately 40 times less lipid soluble than fentanyl) and therefore has slow penetration of the blood-brain barrier.5 In contrast, the high lipid solubility of fentanyl allows rapid penetration of the blood-brain barrier, which leads to a peak effect within several minutes. Conversely, it is this same property (lipid solubility) that allows fentanyl’s rapid redistribution away from the brain and hence its short duration of action. Morphine, with its greater water solubility, crosses the blood-brain barrier much more slowly than fentanyl, resulting in a slower time to peak effect (20 to 30 minutes; Fig. 41-1). Similarly, once morphine has entered the brain, its lower lipid solubility prevents rapid redistribution and hence allows a longer clinical duration of action than fentanyl. Even though morphine appears to be pharmacokinetically similar to fentanyl, it typically has a slower onset time and longer duration of action.
Morphine is metabolized by both the liver and the kidneys. Although the liver is responsible for the majority of its metabolism, 40% is metabolized by the kidneys.15 The major metabolic byproduct of morphine metabolism is morphine-3-glucuronide, which is known to have intrinsic opiate activity roughly one half that of morphine.16 When using morphine in patients with renal failure, the presence of this compound is important because cases of prolonged sedation do occur.17,18
Cardiovascular and Hemodynamic Effects
For many years, morphine has been used in the management of acute myocardial ischemia and infarction pain and for sedation in patients with underlying myocardial disease.19 Morphine has distinct pharmacodynamic and hemodynamic properties that make it advantageous in cardiac patients. Perhaps the most important of these is morphine’s ability to decrease venous and arterial tone.20,21 It appears that the increase in venous capacitance produced by morphine is relatively greater than the decrease in arterial resistance.22 This effect on venous capacitance is dose-related, and with large doses it is possible to decrease preload to the point of hypotension.23 In certain cardiac patients, a modest decrease in preload, as occurs with a dose of intravenous morphine, is desirable. It appears that in the dosages commonly used in the critical care setting, morphine has no direct effect on the inotropic state of the heart.24 An exception to this is the negative inotropic state caused by histamine release when morphine is administered rapidly or when large doses are given (discussed later).5 Finally, the net chronotropic effect of morphine is to slow the heart rate under usual conditions. The exact mechanism by which morphine achieves this action is not certain, but it is thought to involve both a stimulation of the central vagal nucleus and a direct depressive effect on the sinoatrial node.25,26 Morphine has also been observed to secondarily increase heart rate by causing histamine release (discussed later).
Side Effects, Complications, and Toxicity
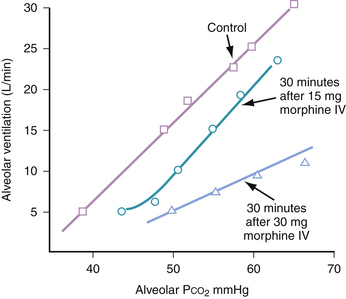
Many of morphine’s side effects are dose-related and can be minimized by reducing the size of doses administered to patients. The most important and potentially dangerous side effect of morphine administration is respiratory depression. The opiate agonists all share the ability to depress ventilation; this is primarily accomplished by decreasing the central ventilatory response to CO2.5 Specifically, the ventilatory response curve to an increasing PaCO2 is shifted to the right, and the slope is decreased (Fig. 41-2). This means that for a given rise in PaCO2, the patient compensates with a less-than-expected minute ventilation rate.5 The typical breathing pattern is one of slow respiratory rate with preserved or even increased tidal volume. This effect is dose-related, and although apnea can occur with high doses, it is usually preceded by a period of progressive hypoventilation and therefore can be identified early and prevented.
The other organ systems most commonly affected by morphine (or any of the opioid agents) are the gastrointestinal (GI) and genitourinary (GU) systems. Morphine has many GI effects, including nausea, emesis, constipation, generalized slowing of the GI tract, and spasm of the sphincter of Oddi. Morphine has been reported to cause urinary retention by increasing urethral sphincter and detrusor tone.27 Hyponatremia secondary to the syndrome of inappropriate secretion of antidiuretic hormone is also occasionally seen with the administration of large doses of morphine.28 When morphine is given to patients with renal failure, prolonged narcotic effect from the accumulation of the active metabolite morphine-3-glucuronide can cause excessive sedation or respiratory depression.17
Although true allergic reactions to morphine are quite rare, morphine is known to cause the release of histamine.29 The release of histamine is from mast cells rather than the basophils, and the mechanism, although not thoroughly understood, is nonimmunologic.30 The release of histamine can lead to a warm flushing sensation, intense pruritus, hypotension, and tachycardia. When hypotension related to histamine release occurs, treatment includes discontinuing morphine infusion, ruling out other causes of anaphylactic or anaphylactoid type of reactions, administering intravenous fluids for hypotension, and administering histamine type 1 and type 2 blocking agents.5 One study has demonstrated that pretreatment with histamine type 1 and type 2 receptor antagonists in patients who have received morphine and tubocurarine (another agent associated with histamine release) significantly decreases hypotension when compared with similar patients pretreated with a placebo, although skin flushing was not reduced.30 Other side effects, including respiratory depression, sphincter of Oddi spasm, excessive sedation, and pruritus, can be reversed by the administration of the opiate antagonist naloxone. By titrating small doses of naloxone, it is possible to reverse the side effects of morphine without completely reversing the analgesia. It is prudent to avoid giving large doses of naloxone to cardiac patients because complete reversal of opiate effect can result in dangerous increases in sympathetic tone and, rarely, has been reported to cause pulmonary edema.31
Fentanyl
History and Structure
Fentanyl and meperidine are probably the best known of the synthetic opioid analgesic agents. These agents, along with sufentanil and alfentanil, are members of the phenylpiperidine class of opiate agents. They are named for the phenylpiperidine skeleton in their chemical structure.5 In terms of analgesic properties, fentanyl is approximately 80 times more potent than morphine because of its greater affinity for the μ-opiate receptor.15
Fentanyl was introduced in 1959 and has been in clinical use since the early 1960s, when it was first used in a combination preparation with the butyrophenone agent droperidol (Innovar) for the technique of neuroleptanesthesia.32,33 For many years, fentanyl was used almost exclusively in the operating room by anesthesiologists because of safety concerns related to its high potency and rapid onset. In the past few decades, fentanyl has found greater use as an analgesic and sedative in the critical care setting.
Pharmacokinetics, Pharmacodynamics, and Metabolism
The initial redistribution half-life for fentanyl is short (1 to 2 minutes), the t1/2α is longer (10 to 30 minutes), and the t1/2β is 2 to 4.5 hours.14 Therefore, pharmacokinetically, fentanyl behaves similarly to morphine; this fact is supported by the nearly identical elimination curve shared by the two drugs. Fentanyl is approximately 40 times more lipid-soluble than morphine, leading to significant differences in the pharmacodynamics between these two drugs. Fentanyl’s great lipid solubility allows rapid entry of this drug into the brain, causing a peak effect minutes after a bolus administration.5 Peak effect parallels the serum level for this agent.14 As a corollary, fentanyl redistributes away from the brain very quickly, resulting in a short clinical duration of action of only 30 to 60 minutes after a bolus dose. For these reasons, fentanyl administered by constant infusion leads to more consistent analgesia.34 Prolonged narcotic effect may be observed with fentanyl after prolonged infusions or frequent bolus dose administration.35 The reason for this prolongation of half-life with long-term infusion of fentanyl is thought to be that the high lipid solubility of this agent allows absorption of large amounts of drug into poorly perfused fatty tissue. When this occurs, termination of effect no longer depends on redistribution but rather is dependent on the terminal half-life of the drug. In a situation analogous to that seen with prolonged infusions of sodium thiopental, fatty uptake leads to a prolonged t1/2β and a long clinical duration of effect.5,36,37
Fentanyl is metabolized primarily by the liver and somewhat by the kidney. Active metabolites of fentanyl are probably of minimal or no clinical significance.15
Cardiovascular and Hemodynamic Effects
When used in analgesic doses, fentanyl has developed a reputation for causing minimal hemodynamic effects. It has been demonstrated that fentanyl does not have negative inotropic effects.38,39 Although its occurrence is unusual, fentanyl can cause hypotension. The etiologies of fentanyl-related hypotension are likely fentanyl-induced bradycardia and a decrease in central sympathetic tone. The cause of the bradycardia is thought to be direct stimulation of the central vagal nucleus by fentanyl.40 Furthermore, the magnitude of the bradycardia is believed to be related to both the total dose administered and the rate of infusion.5,40 Fentanyl is also known to cause hypotension indirectly by decreasing central sympathetic outflow.41 This mechanism is supported by the observation that patients with high basal levels of sympathetic tone are more likely to become hypotensive when given fentanyl.5 Patients with relative or absolute hypovolemia are more prone to hypotension after receiving fentanyl. Although fentanyl administration has minimal hemodynamic effects, it is important to note that reports have suggested that the combination of synthetic opioids and benzodiazepines, especially midazolam, can cause significant decreases in blood pressure.42 It is possible that lorazepam combined with fentanyl may cause less hypotension than does the combination of midazolam and fentanyl.43
Side Effects, Complications, and Toxicity
The side effect and toxicity profile of fentanyl is similar to that of morphine, with several exceptions. The cardiovascular and hemodynamic effects of fentanyl have been discussed previously. The respiratory, central nervous system, GI, and GU side effects are similar to those previously discussed for morphine. The pharmacodynamic properties of fentanyl result in apnea and oversedation quickly; therefore, this drug should be used only in a monitored situation. Fentanyl does not release histamine from mast cells in humans.44 It has been rarely reported to cause true anaphylactic reactions in humans.5 The respiratory, GI, and GU side effects of fentanyl are responsive to treatment with the narcotic antagonist naloxone. The same cautions described for the use of naloxone with morphine also apply to its use with fentanyl.
Benzodiazepines
History and Structure
The benzodiazepines, which are the most commonly used sedative agents in the critical care unit, have been in use for several decades. The first benzodiazepine, chlordiazepoxide, was formulated in 1955, but it was not noted to have sedative properties until 1957 and was not released for clinical use until 1959.45 Chlordiazepoxide was followed in 1959 by the synthesis of diazepam, in 1971 by the formulation of lorazepam, and in 1976 by the synthesis of midazolam.46,47 In addition to its unique pharmacologic properties (discussed later), midazolam is of great interest to intensivists and anesthesiologists in that it is the first benzodiazepine formulated primarily for use in anesthesia and the critical care setting. The benzodiazepines as a group are relatively small, lipid-soluble molecules that act as agonists at the benzodiazepine receptor.46
Site of Action and Receptor Physiology
In 1977, 18 years after their clinical introduction, it was discovered that the site of action of the benzodiazepines was at a receptor complex that it shared with another important class of central nervous system depressants, the barbiturates.48 This receptor, named the BNZ receptor, along with the barbiturate receptor, forms part of a larger receptor system known as the γ-aminobutyric acid (GABA) receptor complex.14 The benzodiazepines act agonistically at this receptor. GABA, one of the primary inhibitory neurotransmitters of the human central nervous system, acts as an agonist at the GABA complex, causing a net influx of chloride ion into the cell, which results in hyperpolarization and resistance to excitation.49 It appears that there are two main GABA receptors in the central nervous system: a GABAa complex and a GABAb complex. The benzodiazepines appear to have their main activity at the GABAa complex.14 When a benzodiazepine binds at its BNZ receptor, the conformation of the GABAa complex is altered such that the binding of the neurotransmitter GABA is facilitated.46 Benzodiazepine receptors are found in greatest concentration in the olfactory bulb, cerebral cortex, cerebellum, hippocampus, substantia nigra, and inferior colliculus.46
Pharmacokinetics, Pharmacodynamics, and Metabolism
In the United States, the three benzodiazepines most widely available for intravenous administration, and hence most commonly used in the CICU, are diazepam, lorazepam, and midazolam. Although these agents work similarly at the receptor level, they are quite different with respect to their pharmacology and physical properties (Table 41-3). For example, these agents differ in potency. Midazolam is roughly three times more potent than diazepam, whereas lorazepam is five times more potent than diazepam.46 In general, all three compounds are highly lipid soluble, but lorazepam is less so than diazepam or midazolam.50 The initial t1/2α of these agents is similar: 1 to 2 minutes for midazolam and diazepam and about 3 minutes for lorazepam.51,52 The onset of peak clinical activity mirrors these half-lives; the slightly slower onset of lorazepam is related to its lower lipid solubility. The t1/2β is quite different among these three drugs and is related to a number of factors (discussed later). The absolute range in t1/2β is large; midazolam has a relatively short half-life of 2 to 3 hours, lorazepam has an intermediate half-life of 10 to 20 hours, and diazepam has a long half-life of 20 to 50 hours.46
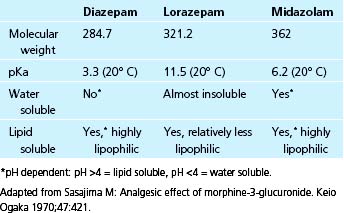
The benzodiazepines are metabolized in the liver where the parent compounds undergo extensive biotransformation. This often results in metabolic products that have significant benzodiazepine activity. Diazepam, the archetypal compound, undergoes biotransformation to a number of products, two of which (oxazepam and desmethyldiazepam) are potent and long-acting BNZ receptor agonists.14 This fact helps to explain the long t1/2β and prolonged sedative effect frequently seen with the use of diazepam.53 Lorazepam is also highly metabolized, but it appears that none of its metabolic compounds have significant activity and that these products are rapidly excreted by the kidneys.51 Midazolam is biotransformed to compounds known as hydroxymidazolams, but controversy exists regarding whether these compounds have any intrinsic benzodiazepine activity.54 As mentioned earlier, there have been many case reports of prolonged sedation from long-term use of diazepam, and this has been traditionally thought to be an effect of accumulation of active metabolites.53 There are reports of prolonged sedation with midazolam infusions. Several studies have shown a prolongation of t1/2β and duration of sedation with midazolam administration in elderly patients and in the critically ill.55,56
Related posts:
 Evolution of the Coronary Care Unit: Past, Present, and Future
Evolution of the Coronary Care Unit: Past, Present, and Future
 Elevated Cardiac Troponin in the Absence of Acute Coronary Syndromes: Mechanism, Significance, and Prognosis
Elevated Cardiac Troponin in the Absence of Acute Coronary Syndromes: Mechanism, Significance, and Prognosis
 Conduction Disturbances in Acute Myocardial Infarction
Conduction Disturbances in Acute Myocardial Infarction
 Pharmacologic Interactions in the CICU
Pharmacologic Interactions in the CICU
 Acute Aortic Syndromes: Diagnosis and Management
Acute Aortic Syndromes: Diagnosis and Management
 Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


