Key Concepts
 Adrenergic agonists can be categorized as direct or indirect. Direct agonists bind to the receptor, whereas indirect agonists increase endogenous neurotransmitter activity.
Adrenergic agonists can be categorized as direct or indirect. Direct agonists bind to the receptor, whereas indirect agonists increase endogenous neurotransmitter activity.
 The primary effect of phenylephrine is peripheral vasoconstriction with a concomitant rise in systemic vascular resistance and arterial blood pressure.
The primary effect of phenylephrine is peripheral vasoconstriction with a concomitant rise in systemic vascular resistance and arterial blood pressure.
 Clonidine seems to decrease anesthetic and analgesic requirements and to provide sedation and anxiolysis.
Clonidine seems to decrease anesthetic and analgesic requirements and to provide sedation and anxiolysis.
 Dexmedetomidine is a lipophylic α-methylol derivative with a higher affinity for α2-receptors than clonidine. It has sedative, analgesic, and sympatholytic effects that blunt many of the cardiovascular responses seen during the perioperative period.
Dexmedetomidine is a lipophylic α-methylol derivative with a higher affinity for α2-receptors than clonidine. It has sedative, analgesic, and sympatholytic effects that blunt many of the cardiovascular responses seen during the perioperative period.
 Long-term use of these agents, particularly clonidine and dexmedetomidine, leads to supersensitization and up-regulation of receptors; with abrupt discontinuation of either drug, an acute withdrawal syndrome manifested by a hypertensive crisis can occur.
Long-term use of these agents, particularly clonidine and dexmedetomidine, leads to supersensitization and up-regulation of receptors; with abrupt discontinuation of either drug, an acute withdrawal syndrome manifested by a hypertensive crisis can occur.
 Ephedrine is commonly used as a vasopressor during anesthesia. As such, its administration should be viewed as a temporizing measure while the cause of hypotension is determined and remedied.
Ephedrine is commonly used as a vasopressor during anesthesia. As such, its administration should be viewed as a temporizing measure while the cause of hypotension is determined and remedied.
 Small doses (approximately 2 mcg/kg/min) of dopamine (DA) have minimal adrenergic effects but activate dopaminergic receptors. Stimulation of these nonadrenergic receptors (specifically, DA1 receptors) vasodilates the renal vasculature and promotes diuresis.
Small doses (approximately 2 mcg/kg/min) of dopamine (DA) have minimal adrenergic effects but activate dopaminergic receptors. Stimulation of these nonadrenergic receptors (specifically, DA1 receptors) vasodilates the renal vasculature and promotes diuresis.
 Favorable effects on myocardial oxygen balance are believed to make dobutamine a good choice for patients with the combination of congestive heart failure and coronary artery disease, particularly if peripheral vascular resistance is elevated. (There are some recent debates regarding this beneficial effect.)
Favorable effects on myocardial oxygen balance are believed to make dobutamine a good choice for patients with the combination of congestive heart failure and coronary artery disease, particularly if peripheral vascular resistance is elevated. (There are some recent debates regarding this beneficial effect.)
 Labetalol lowers blood pressure without reflex tachycardia because of its combination of α- and β-effects.
Labetalol lowers blood pressure without reflex tachycardia because of its combination of α- and β-effects.
 Esmolol is an ultrashort-acting selective β1-antagonist that reduces heart rate and, to a lesser extent, blood pressure.
Esmolol is an ultrashort-acting selective β1-antagonist that reduces heart rate and, to a lesser extent, blood pressure.
 Discontinuation of β-blocker therapy for 24-48 hr may trigger a withdrawal syndrome characterized by hypertension, tachycardia, and angina pectoris.
Discontinuation of β-blocker therapy for 24-48 hr may trigger a withdrawal syndrome characterized by hypertension, tachycardia, and angina pectoris.
Adrenergic Agonists & Antagonists: Introduction
Adrenergic agonists and antagonists produce their clinical effects by interacting with the adrenergic receptors (ie, adrenoceptors). The clinical effects of these drugs can be deduced from an understanding of the adrenoceptor physiology and a knowledge of which receptors each drug activates or blocks.
The term “adrenergic” originally referred to the effects of epinephrine (adrenaline), although norepinephrine (noradrenaline) is the primary neurotransmitter responsible for most of the adrenergic activity of the sympathetic nervous system. With the exception of eccrine sweat glands and some blood vessels, norepinephrine is released by postganglionic sympathetic fibers at end-organ tissues (Figure 14-1). In contrast, acetylcholine is released by preganglionic sympathetic fibers and all parasympathetic fibers.
Figure 14-1
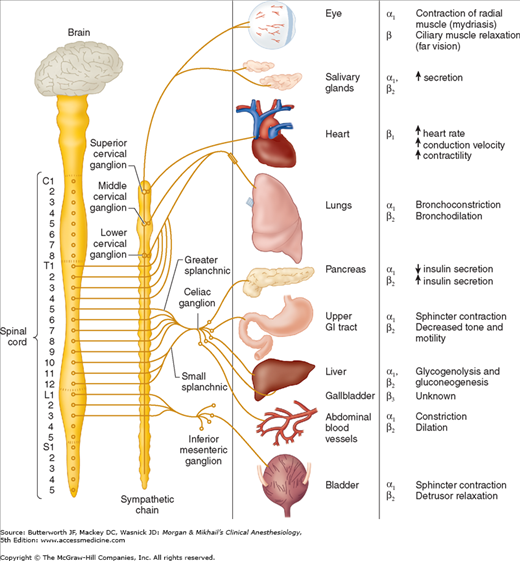
The sympathetic nervous system. Organ innervation, receptor type, and response to stimulation. The origin of the sympathetic chain is the thoracoabdominal (T1-L3) spinal cord, in contrast to the craniosacral distribution of the parasympathetic nervous system. Another anatomic difference is the greater distance from the sympathetic ganglion to the visceral structures.
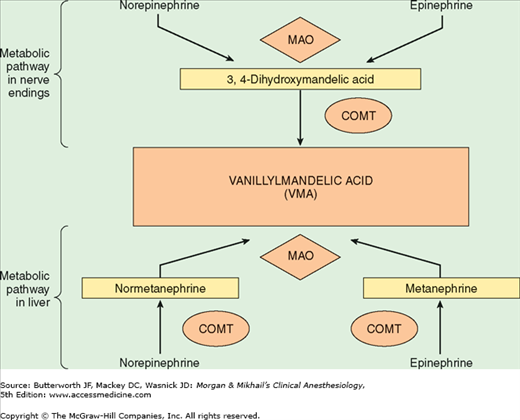
Norepinephrine is synthesized in the cytoplasm of sympathetic postganglionic nerve endings and stored in the vesicles (Figure 14-2). After release by a process of exocytosis, the action of norepinephrine is primarily terminated by reuptake into the postganglionic nerve ending (inhibited by tricyclic antidepressants), but also by diffusion from receptor sites, or via metabolism by monoamine oxidase (inhibited by monoamine oxidase inhibitors) and catechol-O-methyltransferase (Figure 14-3). Prolonged adrenergic activation leads to desensitization and hyporesponsiveness to further stimulation.
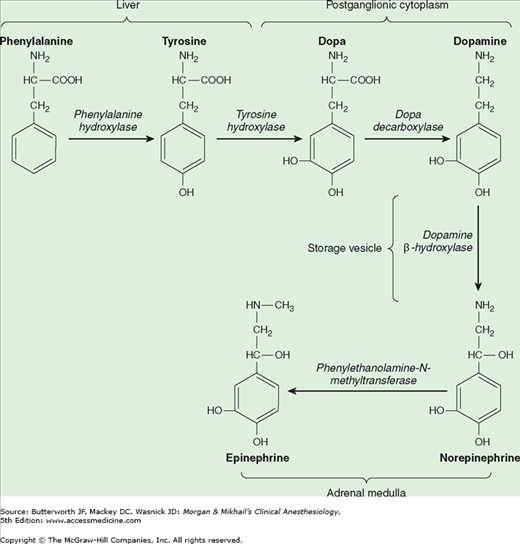
Adrenergic receptors are divided into two general categories: α and β. Each of these has been further subdivided into at least two subtypes: α1 and α2, and β1, β2, and β3. The α-receptors have been further divided using molecular cloning techniques into α1A, α1B, α1D, α2A, α2B, and α2C. These receptors are linked to G proteins (Figure 14-4; Drs. Rodbell and Gilman received the Nobel Prize in physiology or medicine in 1994 for their discovery)—heterotrimeric receptors with α, β, and γ subunits. The different adrenoceptors are linked to specific G proteins, each with a unique effector, but each using guanosine triphosphate (GTP) as a cofactor. α1 is linked to Gq, which activates phospholipases; α2 is linked to Gi, which inhibits adenylate cyclase, and β is linked to Gs, which activates adenylate cyclase.
Figure 14-4
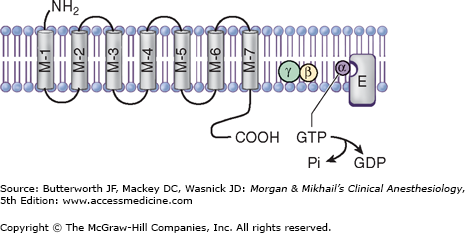
The adrenoceptor is a transmembrane-spanning receptor made up of seven subunits, which is linked to a G protein. G proteins are trimeric endoplasmic membrane proteins made of α, β, and γ units. With activation, GTP on the α-subunit is replaced by GDP, stimulating a conformational change, disassociating the α, β, and γ units. Either the Gα or Gβγ subunits can activate (or inhibit) the enzyme effector for that adrenoceptor. M1-M7, membrane-spanning units; α, β, γ, subunits of G protein; GTP, guanosine triphosphate; Pi, inorganic phosphate—quickly assimilated; GDP, guanosine diphosphate; E, effector; cyclophosphatase for Gq, adenylate cyclase for Gi, and Gs.
α1-Receptors are postsynaptic adrenoceptors located in smooth muscle throughout the body (in the eye, lung, blood vessels, uterus, gut, and genitourinary system). Activation of these receptors increases intracellular calcium ion concentration, which leads to contraction of smooth muscles. Thus, α1-agonists are associated with mydriasis (pupillary dilation due to contraction of the radial eye muscles), bronchoconstriction, vasoconstriction, uterine contraction, and constriction of sphincters in the gastrointestinal and genitourinary tracts. α1-stimulation also inhibits insulin secretion and lipolysis. The myocardium possesses α1-receptors that have a positive inotropic effect, which might play a role in catecholamine-induced arrhythmia. During myocardial ischemia, enhanced α1-receptor coupling with agonists is observed. Nonetheless, the most important cardiovascular effect of α1-stimulation is vasoconstriction, which increases peripheral vascular resistance, left ventricular afterload, and arterial blood pressure.
In contrast to α1-receptors, α2-receptors are located primarily on the presynaptic nerve terminals. Activation of these adrenoceptors inhibits adenylate cyclase activity. This decreases the entry of calcium ions into the neuronal terminal, which limits subsequent exocytosis of storage vesicles containing norepinephrine. Thus, α2-receptors create a negative feedback loop that inhibits further norepinephrine release from the neuron. In addition, vascular smooth muscle contains postsynaptic α2-receptors that produce vasoconstriction. More importantly, stimulation of postsynaptic α2-receptors in the central nervous system causes sedation and reduces sympathetic outflow, which leads to peripheral vasodilation and lower blood pressure.
β-Adrenergic receptors are classified into β1, β2, and β3 receptors. The catecholamines, norepinephrine, and epinephrine are equipotent on β1 receptors, but epinephrine is significantly more potent than norepinephrine on β2 receptors.
The most important β1-receptors are located on the postsynaptic membranes in the heart. Stimulation of these receptors activates adenylate cyclase, which converts adenosine triphosphate to cyclic adenosine monophosphate and initiates a kinase phosphorylation cascade. Initiation of the cascade has positive chronotropic (increased heart rate), dromotropic (increased conduction), and inotropic (increased contractility) effects.
β2-Receptors are primarily postsynaptic adrenoceptors located in smooth muscle and gland cells. They share a common mechanism of action with β1-receptors: adenylate cyclase activation. Despite this commonality, β2 stimulation relaxes smooth muscle, resulting in bronchodilation, vasodilation, and relaxation of the uterus (tocolysis), bladder, and gut. Glycogenolysis, lipolysis, gluconeogenesis, and insulin release are stimulated by β2-receptor activation. β2-agonists also activate the sodium-potassium pump, which drives potassium intracellularly and can induce hypokalemia and dysrhythmias.
β3-Receptors are found in the gallbladder and brain adipose tissue. Their role in gallbladder physiology is unknown, but they are thought to play a role in lipolysis and thermogenesis in brown fat.
Dopamine (DA) receptors are a group of adrenergic receptors that are activated by dopamine; these receptors are classified as D1 and D2 receptors. Activation of D1 receptors mediates vasodilation in the kidney, intestine, and heart. D2 receptors are believed to play a role in the antiemetic action of droperidol.
Adrenergic Agonists
Adrenergic agonists interact with varying specificity (selectivity) at α- and β-adrenoceptors (Tables 14-1 and 14-2).
| Drug | α1 | α2 | β1 | β2 | DA1 | DA2 |
|---|---|---|---|---|---|---|
| Phenylephrine | +++ | + | 0 | 0 | 0 | 0 |
| Methyldopa | + | + | 0 | 0 | 0 | 0 |
| Clonidine | + | ++ | 0 | 0 | 0 | 0 |
| Dexmedetomidine | + | +++ | 0 | 0 | 0 | 0 |
| Epinephrine2 | ++ | ++ | +++ | ++ | 0 | 0 |
| Ephedrine3 | ++ | ? | ++ | + | 0 | 0 |
| Fenoldopam | 0 | 0 | 0 | 0 | +++ | 0 |
| Norepinephrine2 | ++ | ++ | ++ | 0 | 0 | 0 |
| Dopamine2 | ++ | ++ | ++ | + | +++ | +++ |
| Dopexamine | 0 | 0 | + | +++ | ++ | +++ |
| Dobutamine | 0/+ | 0 | +++ | + | 0 | 0 |
| Terbutaline | 0 | 0 | + | +++ | 0 | 0 |
| Drug | Heart Rate | Mean Arterial Pressure | Cardiac Output | Peripheral Vascular Resistance | Bronchodilation | Renal Blood Flow |
|---|---|---|---|---|---|---|
| Phenylephrine | ↓ | ↑↑↑ | ↓ | ↑↑↑ | 0 | ↓↓↓ |
| Epinephrine | ↑↑ | ↑ | ↑↑ | ↑/↓ | ↑↑ | ↓↓ |
| Ephedrine | ↑↑ | ↑↑ | ↑↑ | ↑ | ↑↑ | ↓↓ |
| Fenoldopam | ↑↑ | ↓↓↓ | ↓/↑ | ↓↓ | 0 | ↑↑↑ |
| Norepinephrine | ↓ | ↑↑↑ | ↓/↑ | ↑↑↑ | 0 | ↓↓↓ |
| Dopamine | ↑/↑↑ | ↑ | ↑↑↑ | ↑ | 0 | ↑↑↑ |
| Dopexamine | ↑/↑↑ | ↓/↑ | ↑↑ | ↑ | 0 | ↑ |
| Isoproterenol | ↑↑↑ | ↓ | ↑↑↑ | ↓↓ | ↑↑↑ | ↓/↑ |
| Dobutamine | ↑ | ↑ | ↑↑↑ | ↓ | 0 | ↑ |
Overlapping of activity complicates the prediction of clinical effects. For example, epinephrine stimulates α1-, α2-, β1-, and β2-adrenoceptors. Its net effect on arterial blood pressure depends on the balance between α1-vasoconstriction, α2– and β2-vasodilation, and β1-inotropic influences. Moreover, this balance changes at different doses.
 Adrenergic agonists can be categorized as direct or indirect. Direct agonists bind to the receptor, whereas indirect agonists increase endogenous neurotransmitter activity. Mechanisms of indirect action include increased release or decreased reuptake of norepinephrine. The differentiation between direct and indirect mechanisms of action is particularly important in patients who have abnormal endogenous norepinephrine stores, as may occur with use of some antihypertensive medications or monoamine oxidase inhibitors. Intraoperative hypotension in these patients should be treated with direct agonists, as their response to indirect agonists will be altered.
Adrenergic agonists can be categorized as direct or indirect. Direct agonists bind to the receptor, whereas indirect agonists increase endogenous neurotransmitter activity. Mechanisms of indirect action include increased release or decreased reuptake of norepinephrine. The differentiation between direct and indirect mechanisms of action is particularly important in patients who have abnormal endogenous norepinephrine stores, as may occur with use of some antihypertensive medications or monoamine oxidase inhibitors. Intraoperative hypotension in these patients should be treated with direct agonists, as their response to indirect agonists will be altered.
Another feature distinguishing adrenergic agonists from each other is their chemical structure. Adrenergic agonists that have a 3,4-dihydroxybenzene structure (Figure 14-5) are known as catecholamines. These drugs are typically short-acting because of their metabolism by monoamine oxidase and catechol-O-methyltransferase. Patients taking monoamine oxidase inhibitors or tricyclic antidepressants may therefore demonstrate an exaggerated response to catecholamines. The naturally occurring catecholamines are epinephrine, norepinephrine, and DA. Changing the side-chain structure (R1, R2, R3) of naturally occurring catecholamines has led to the development of synthetic catecholamines (eg, isoproterenol and dobutamine), which tend to be more receptor specific.
Figure 14-5

Adrenergic agonists that have a 3,4-dihydroxybenzene structure are known as catecholamines. Substitutions at the R1, R2
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


