CHAPTER 27 Acute Presentations of Valvular Heart Disease
Acute Aortic Insufficiency
Etiology
Aortic insufficiency occurs as a result of either dilation of the aortic root and annulus or disruption of the valve leaflets. The most common etiologies of acute aortic insufficiency are infective endocarditis and aortic dissection.1 Infective endocarditis is more likely to occur on a congenitally abnormal or rheumatically involved valve. Infective endocarditis results in acute aortic insufficiency through a process of endothelial damage, development of nonbacterial thrombotic vegetation, adherence of circulating organisms to the vegetation, proliferation of infection within the vegetation, and valve destruction.2 Acute, type A, aortic dissection is complicated by some degree of aortic insufficiency in approximately 50% of cases.3,4 Abnormal wall stress in the presence of aortic media disease, such as that seen with chronic hypertension or Marfan disease, results in an intimal tear and subsequent dissection.5 Other etiologies of acute aortic insufficiency are listed in Table 27-1.
Table 27–1 Etiologies of Acute Aortic Insufficiency
| Infective endocarditis |
| Aortic dissection—predisposing and associated conditions |
| Hypertension |
| Marfan syndrome |
| Congenital bicuspid aortic valve |
| Coarctation of aorta |
| Ehler-Danlos syndrome |
| Turner syndrome |
| Chest trauma |
| Rupture of a myxomatous valve |
| Systemic connective tissue disorders |
| Ankylosing spondylitis |
| Systemic lupus erythematosus |
| Granulomatous diseases |
| Tertiary syphilis |
| Giant cell arteritis |
| Takayasu arteritis |
Pathophysiology
In acute, severe, aortic insufficiency, the large regurgitant volume imposed on the unprepared ventricle markedly reduces forward stroke volume and shifts the LV diastolic pressure-volume relationship to the steep ascending portion of the curve. Because the LV has limited distensibility, the acute increase in LV end-diastolic volume due to regurgitant flow results in an abrupt rise in LV end-diastolic pressure (LVEDP) (Fig. 27-1) and, subsequently, pulmonary venous pressure. In addition, reflex sympathetic activation, in response to a reduction in cardiac output and systemic blood pressure, produces tachycardia and increases systemic vascular resistance (SVR). This rise in SVR further worsens regurgitant flow and impedes LV ejection so that a rise in aortic systolic pressure is inhibited. As opposed to chronic aortic regurgitation, aortic diastolic pressure does not fall significantly for two reasons: (1) the rapid increase in LVEDP reduces the driving gradient between aorta and LV and (2) peripheral runoff is limited by an increase in SVR.6,7 In some cases, the LV and aortic diastolic pressures are equalized.
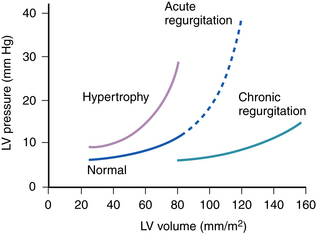
In contrast, the gradual progression of chronic aortic insufficiency allows the ventricle to dilate and undergo eccentric hypertrophy, which shifts the LV pressure-volume curve rightward (see Fig. 27-1), and maintains a normal LVEDP. This compensatory mechanism also increases total stroke volume to preserve forward stroke volume despite the presence of a large regurgitant volume. As a result, there is no reflex tachycardia or rise in SVR. Aortic systolic pressure rises as a result of the increased stroke volume, and diastolic pressure falls in response to the regurgitant volume, decreased SVR, and rapid peripheral runoff. These factors are responsible for the classic finding of a wide pulse pressure in chronic aortic insufficiency.
Clinical Presentation
The clinical features of acute aortic insufficiency are profoundly different from those of chronic disease. These differences result primarily from the presence of markedly elevated LVEDP and absence of a wide pulse pressure in patients with acute severe aortic insufficiency as described above. Because the pulse pressure is usually normal in the acute setting, the classic findings of severe aortic insufficiency in the periphery are absent (Table 27-2). In addition, the striking elevation in LVEDP accounts for the often dramatic presentation of congestive heart failure and cardiogenic shock.8
Table 27–2 Classic Peripheral Manifestations of Chronic Aortic Insufficiency∗
| Head bobbing with each systole |
| Water-hammer pulse felt with abrupt distention and quick collapse |
| “Pistol shot” sounds—booming systolic and diastolic sounds heard over the femoral artery |
| Systolic pulsations of the uvula |
| Systolic murmur heard over the femoral artery when it is compressed proximally and diastolic murmur when it is compressed distally |
| Visible capillary pulsations in the nail bed (seen by shining light source), fingertips, or lip (detected with glass slide pressed against the lip) |
| Popliteal cuff systolic pressure exceeding brachial cuff pressure by more than 60 mm Hg |
∗ Not typically seen with acute aortic insufficiency.
Patients typically have severe dyspnea, weakness, or hypotension. They are often tachycardic, and the LV impulse may be normal. Early mitral valve closure, a hallmark of severe acute aortic insufficiency, results from a rapid elevation of LVEDP, which exceeds left atrial (LA) pressure early in diastole. This reversal of pressures between the LV and LA in late diastole results in a soft or inaudible first heart sound (S1). Occasionally, mitral valve closure may be heard during diastole and accompanied by diastolic mitral regurgitation.9,10 The Austin-Flint murmur, which is thought to represent turbulent flow from the LA to the LV because of partial mitral valve closure from the aortic insufficiency jet is either absent or brief and ceases when LV pressure exceeds LA pressure in diastole.11,12 An accentuated pulmonic closure sound suggests pulmonary hypertension, and a third heart sound (S3) is frequently heard. A fourth heart sound (S4), however, is usually not present because the mitral valve is either closed before atrial systole occurs or LVEDP is already so high that there is little flow to the ventricle during this period. The acute aortic insufficiency murmur is usually short, early, and of medium pitch, which is in contrast to the long, high-pitched murmur of chronic aortic insufficiency. In tachycardic patients, this murmur can easily be overlooked. Edema and weight gain are not often seen in severe acute aortic insufficiency because there is inadequate time for substantial secondary salt and water retention. The extremities may be cool and mottled, owing to both poor cardiac output and elevated SVR.
Diagnosis
An ECG is important in the evaluation of any patient with pulmonary edema, primarily to rule out myocardial infarction. In the absence of pre-existing heart disease, the chest radiograph generally reveals a normal cardiac silhouette with evidence of pulmonary edema (Fig. 27-2). Noninvasive imaging by TTE provides crucial information regarding the presence, severity, and etiology of aortic insufficiency. With severe aortic regurgitation, in addition to visualizing the regurgitant jet with color Doppler, quantitative measurements, such as jet or vena contracta (narrowest portion of regurgitant jet just distal to valve orifice) width, can be obtained. A jet width greater than 65% of LV outflow tract and vena contracta greater than 0.6 cm are consistent with severe aortic regurgitation.12A Continuous wave Doppler is used to calculate the pressure half-time, which reflects the equilibration between aortic and LV diastolic pressure. With acute, severe aortic regurgitation, the rapid equilibration of pressures results in a short pressure half-time of less than 200 msec.13 Other echocardiographic findings supportive of severe aortic regurgitation include premature closure of the mitral valve, detected best by M-mode echocardiography and holodiastolic flow reversal in the descending aorta. Transesophageal echocardiography (TEE) may be required in individuals in whom transthoracic echo windows are limited (Fig. 27-3). In addition, TEE has increased sensitivity for evaluating the underlying etiology of aortic regurgitation, such as endocarditis (vegetations or aortic root abscess) or aortic dissection (dissection flap).14,15
Aortic dissection must be considered in the differential diagnosis of any patient having acute aortic regurgitation. This diagnosis is confirmed either by computed tomography (CT), TEE, or magnetic resonance imaging (MRI). These imaging modalities have largely replaced aortography, the previous gold standard. A recent meta-analysis compared the effectiveness of TEE, CT, and MRI for the diagnosis of aortic dissection. After pooling 16 studies with 1139 patients, TEE, helical CT, and MRI were found to have sensitivities of 98%, 100%, and 98%, respectively, and specificities of 95%, 98%, and 98%, respectively.16 CT is the most widely used modality given its speed and ease of accessibility to most emergency rooms (Fig. 27-4). CT also may provide identification of the intimal flap and involvement of major branch arteries. TEE has an advantage in that it is a bedside procedure and allows direct visualization of the aortic regurgitant jet. However, it is unable to detect dissections localized to the distal ascending aorta or proximal aortic arch. MRI has the best positive likelihood ratio for the detection of aortic dissections, but is time consuming, often not readily available in an emergent setting, and not feasible for a critically ill patient.
Treatment
In general, most patients with acute severe aortic insufficiency are desperately ill and have evidence of both systemic hypoperfusion and pulmonary edema. Acute aortic regurgitation usually requires urgent surgery. However, medical therapy has an important role in optimizing hemodynamics perioperatively. In the presence of severe hemodynamic compromise, admission to the cardiac intensive care unit is clearly indicated. The principles of management are to recognize the degree of hemodynamic impairment, reduce pulmonary venous pressure, maximize cardiac output, and initiate therapy for any underlying disorder.17 Invasive hemodynamic monitoring, by placement of a Swan-Ganz pulmonary artery catheter, allows the clinician to assess the response to therapy and gauge the tempo of the illness.
Intravenous vasodilator therapy significantly reduces pulmonary artery pressures. Nitroprusside is the vasodilator of choice, starting at 0.25 μg/kg/min given intravenously and gradually titrating upward by increments of 0.25 to 0.5 μg/kg/min with the goal of achieving optimal hemodynamics or until systemic hypotension supervenes.18 The speed of uptitration is dictated by the degree of hemodynamic compromise. In severely ill patients the nitroprusside dose can be increased every 5 minutes, whereas in stable patients, a more gradual approach can be used. During maintenance therapy, one needs to be alert for signs and symptoms of both cyanide and thiocyanate toxicity. These compounds are breakdown products of nitroprusside, which accumulate with prolonged use, especially in the presence of renal insufficiency.
In general, inotropic agents do not have a significant role in management because most cases of acute aortic insufficiency occur in the setting of normal or even accentuated LV function. However, if pre-existing myocardial dysfunction exists, agents such as dobutamine at a dose of 5 to 15 μg/kg/min may assist in maintaining cardiac output.19 Intra-aortic balloon pumps (IABP) are contraindicated with aortic regurgitation. Additional medical therapy includes appropriate antibiotics in suspected infective endocarditis.20 In the case of aortic dissection, intravenous ß-blockers are thought to be useful in reducing the velocity of LV ejection, thereby minimizing aortic wall stress. However, when aortic dissection is complicated by acute aortic regurgitation, ß-blockers should be used cautiously, if at all, as the compensatory tachycardia would be blunted, further reducing cardiac output.
If, despite medical therapy, hemodynamic instability persists, emergent surgical valve repair or replacement represents the only definitive option for cure. Indications for surgery in the presence of infective endocarditis are outlined in Table 27-3. Even in the presence of active infective endocarditis, valve surgery should not be delayed to achieve bacteriologic cure. In the International Registry of Acute Aortic Dissection, patients with type A dissection had an in-hospital mortality of 26% with surgery and 58% with medical management.21 Of those undergoing surgery for type A aortic dissection, 16% had involvement of the aortic valve requiring valve repair or replacement in conjunction with aortic root/arch surgery.
Table 27–3 Indications for Surgery in Infective Endocarditis
| Native Valve Endocarditis |
| Congestive heart failure refractory to routine management (Class I) |
| Elevated LV end-diastolic pressure or LA pressures (Class I) |
| Fungal or highly-resistant organisms (Class I) |
| Complications: heart block, annular or aortic abscess, destructive penetration lesions (Class I) |
| Recurrent emboli and persistent vegetations despite appropriate antimicrobial therapy (Class IIa) |
| Mobile vegetations is in excess of 10 mm (Class IIb) |
| Prosthetic Valve Endocarditis |
| Congestive heart failure (Class I) |
| Valve dehiscence (Class I) |
| Increasing valve obstruction or worsening regurgitation (Class I) |
| Complications, as above (Class I) |
| Persistent bacteremia or recurrent emboli (Class IIa) |
| Relapsing infection (Class IIa) |
From Bonow RO, Carabello BA, Chatterjee K, et al: ACC/AHA 2006 practice guidelines for the management of patients with valvular heart disease. J Am Coll Cardiol 2006;48:1.
Acute Mitral Regurgitation
Etiology
The presentation of acute severe mitral regurgitation is not unlike acute aortic insufficiency. Both valve lesions result in sudden, severe LV volume overload. To better understand the underlying pathophysiologic states leading to acute mitral regurgitation, it is important to first recognize the functional components of the mitral valve apparatus. These components include the LA, mitral annulus, mitral valve leaflets, chordae tendineae, papillary muscles, and the LV wall. All these structures must work in concert to produce effective mitral valve leaflet apposition during systole.
Infective endocarditis may cause acute mitral regurgitation by mechanisms including leaflet perforation, alteration of mitral valve annulus secondary to abscess formation, or chordae tendineae rupture. Other etiologies of chordal rupture are myxomatous degeneration secondary to mitral valve prolapse or Marfan’s disease spontaneous rupture, trauma, rheumatic disease, or spontaneous.22,23 Coronary artery disease is another major cause of acute mitral regurgitation and may affect valvular function in a number of ways: (1) papillary muscle rupture after myocardial infarction24; (2) ischemic papillary muscle dysfunction25; (3) papillary muscle fibrosis25; (4) dyssynergy of the LV segment adjacent to a normally functioning papillary muscle26; and (5) diffuse LV enlargement with mitral annular dilation. The posteromedial papillary muscle has only one vascular supply arising from either the right coronary or left circumflex artery and is, therefore, more susceptible to ischemic dysfunction or myocardial infarction. With the increasing use of percutaneous balloon valvotomy for rheumatic mitral stenosis, iatrogenic mitral regurgitation requiring valve replacement is more frequent as compared with closed surgical valvotomy.27–29 Finally, degeneration of a bioprosthetic valve, impaired closure of a mechanical valve, or paravalvular regurgitation from suture disruption may lead to acute, prosthetic valve, mitral regurgitation.
Pathophysiology
The severity of mitral regurgitation depends on the volume of regurgitant flow, LA compliance, and pre-existing LV function. The volume of regurgitant flow is a function of the size of the incompetent valve orifice and the pressure gradient between the LV and LA.30 In the presence of a relatively noncompliant LA, the abrupt increase in pressure is transmitted to the pulmonary circuit with resultant pulmonary edema.31 With continuing acute regurgitation, the LV begins to fail secondary to elevated wall stress due to the mismatch between elevated LV end-diastolic volume/pressure and wall mass. In the presence of mitral regurgitation, there are two outlets to flow from the LV: (1) the relatively high-impedance systemic circulation and (2) the low-impedance LA. In this setting, forward stroke volume is highly dependent on SVR. As SVR increases, a greater proportion of the total LV stroke volume is directed to the LA and the regurgitant fraction increases [(total stroke volume − forward stroke volume)/total stroke volume)].32 Thus the reduction in cardiac output increases SVR through neurohormonal activation, which worsens the severity of mitral regurgitation. As regurgitant flow increases further and cardiac output continues to decline, intense peripheral vasoconstriction ensues, leading to a vicious cycle of worsening mitral regurgitation and further impairment in forward stroke volume.
Clinical Presentation
Most patients with acute mitral regurgitation are tachycardic, which represents a compensatory mechanism to maintain cardiac output in the presence of declining forward stroke volume. The jugular venous pulse may be elevated with 50% of patients having a prominent “a” wave.33 Precordial examination often reveals a hyperdynamic, nondisplaced apical impulse with a prominent presystolic expansion suggesting LV overload with increased atrial systole. A left parasternal lift is also common and is an indication of severe mitral regurgitation, often in association with elevated right ventricular systolic pressures. A systolic apical thrill may be felt in up to 75% of patients with ruptured chordae tendineae.33 The presence of a thrill is less common in papillary muscle dysfunction or rupture.34
Cardiac auscultation reveals a normal S1 because in most cases of acute mitral regurgitation the mitral valve leaflets are normal. This is in contradiction to chronic mitral regurgitation in which S1 is soft secondary to intrinsically abnormal mitral valve leaflets. Accentuated pulmonary valve closure suggests pulmonary hypertension33 and because the LV empties rapidly, the aortic component may close early, giving rise to a widened split.35 The presence of an S4 is common. An S3 gallop is almost universally heard and is related to LV volume overload. The murmur of acute mitral regurgitation differs according to the underlying pathophysiology. In papillary muscle dysfunction, a crescendo-decrescendo murmur may be heard during mid-to-late systole, while papillary muscle rupture results in a pansystolic murmur. Acute chordal rupture results in an ejection murmur that begins in the apex and radiates to the base of the heart.36 Chronic mitral regurgitation, on the other hand, gives rise to a soft blowing holosystolic murmur heard throughout systole that begins at the apex and radiates to the axilla and back. Early termination of the murmur in acute mitral regurgitation results from rapid equalization of LA and LV pressures and suggests a greater degree of regurgitation.37 The severity of the murmur may not reflect the degree of volume overload.38 A summary of the differences in clinical presentation between acute and chronic mitral regurgitation is listed in Table 27-4.
Table 27–4 Clinical Features of Severe Mitral Regurgitation
| Feature | Acute | Chronic |
|---|---|---|
| Congestive heart failure | Rapid and sudden | Insidious |
| Rhythm | Sinus tachycardia | Atrial fibrillation |
| Point of maximal impulse | Hyperdynamic and nondisplaced | Hyperdynamic and shifted inferolaterally |
| Right ventricular lift | Present | Absent |
| Precordial thrill | Usually present | Absent |
| Jugular venous pressure | Prominent “a” wave | Normal tracing |
| Heart sounds | ||
| S1 | Normal | Soft |
| S2 | Accentuated P2 with wide split | Normal P2 with wide split |
| S3 | Present | Present |
| S4 | Present | Absent |
| Mitral regurgitation murmur | Loud, decreasing in late systole | Blowing holosystolic |
| Radiation of mitral regurgitation murmur | Toward base | Toward axilla |
| Mitral diastolic flow murmur | Present | Absent |
From Depace NL, Nestico PF, Morganroth J: Acute severe mitral regurgitation: pathophysiology, clinical recognition and management. Am J Med 1985;78:293.
Diagnosis
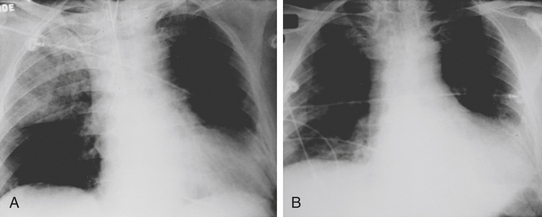
As in the case of acute aortic insufficiency, immediate assessment of intracardiac filling pressures becomes critical, especially in the patient who is hemodynamically unstable. Initial noninvasive diagnostic maneuvers include a chest radiograph, which typically reveals a normal cardiac silhouette with pulmonary venous congestion or edema.39 However, with pre-existing valvular or myocardial disease, there may be radiographic evidence of cardiac enlargement. Occasionally, an unusual pattern of right upper lobe pulmonary edema40 may result that can be confused with pneumonia. However, prompt resolution with diuretic and vasodilator therapy rapidly clarifies the diagnosis (Fig. 27-5). TEE has demonstrated that this radiologic finding may be related to the regurgitant jet being directed toward the right superior pulmonary vein.41
Because the underlying pathoanatomy of the mitral valve influences prognosis and determines the type of therapeutic intervention, echocardiographic assessment of the mitral valve apparatus becomes essential. In the presence of good echocardiographic windows, transthoracic imaging can be performed quickly and safely at the bedside to determine the underlying etiology and severity of mitral regurgitation. In addition, overall LV function and wall motion abnormalities indicative of ischemia or infarction can be assessed. Finally, structural cardiac disorders that mimic mitral regurgitation, such as ventricular septal rupture, can be ruled out.42
Depending on the etiology of acute mitral regurgitation, a variety of echocardiographic abnormalities may be seen. There may be an obvious flail leaflet, chordal rupture, or vegetation. Papillary muscle rupture is often directly visualized as a mass attached to the involved leaflet with discontinuity of the base of the muscle.43 Despite the accuracy of transthoracic imaging, technical difficulties may impair visualization and limit interpretation. In these circumstances, TEE is a useful alternative modality for assessing acute mitral regurgitation (Fig. 27-6). Compared with TTE, TEE has superior resolution and a significant advantage in terms of visualizing the mitral valve apparatus, especially in the presence of a prosthetic mitral valve. Doppler imaging provides both qualitative and quantitative assessment of the mitral regurgitation severity. A color Doppler jet width at the vena contracta of more than 6 mm by multiplane TEE identifies angiographically severe mitral regurgitation with a sensitivity and specificity of 95% and 98%, respectively.44 If systolic retrograde flow into the pulmonary veins is detected, mitral regurgitation is at least moderate. Finally, echocardiography clearly distinguishes acute mitral regurgitation from ventricular septal rupture, which has a very similar clinical presentation (Table 27-5).
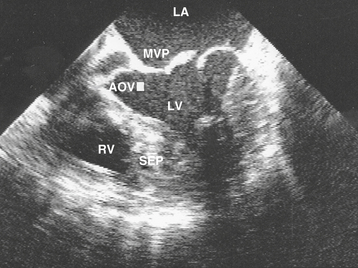
Table 27–5 Differentiation of Papillary Muscle Rupture and Ventricular Septal Rupture
| Feature | Papillary Muscle Rupture | Ventricular Septal Rupture |
|---|---|---|
| Age (mean, years) | 65 | 63 |
| Days post myocardial infarction | 3-5 | 3-5 |
| Anterior myocardial infarction | 25% | 66% |
| Murmur | Variable systolic | Pansystolic at lower sternal border |
| Palpable thrill | Rare | Yes |
| “v” wave in pulmonary capillary wedge tracing | + + | + + |
| Oxygen step-up from right atrium to pulmonary artery | ±∗ | + + |
| Echocardiographic findings | Flail or prolapsing leaflet | Visualize defect |
| Doppler | Regurgitant jet in LA | Detect shunt |
| Mortality | ||
| Medical | 90% | 90% |
| Surgical | 40%-90% | 50% |
+ +, invariably present; +, occasionally present; ±, rarely present.
∗ Oxygen step-up may occasionally be seen in papillary muscle rupture as a result of the regurgitant “v” from left atrium contaminating the mixed venous sample from the pulmonary artery.
From Antman EM: ST-elevation myocardial infarction: management. In Zipes DP, Libby P, Bonow RO, et al (eds): Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine, 7th ed. Philadelphia, WB Saunders, 2005, p 1204.
If diagnostic studies, including ECG and echocardiography indicate ischemia or infarction as the underlying etiology of acute mitral regurgitation, then urgent cardiac catheterization must be considered depending on the hemodynamic stability of the patient. Coronary angiography defines coronary anatomy and may delineate a culprit lesion amenable to catheter-based or surgical intervention.
Treatment
The management of acute severe mitral regurgitation is similar to that of acute aortic insufficiency. The principles of treatment focus on reducing LVEDP, decreasing aortic impedance to LV ejection so that blood flow can be directed in a forward rather than retrograde direction, and initiating specific therapy for the precipitating etiology. As with acute aortic insufficiency, right heart catheterization is an integral component in the management of acute mitral regurgitation. The clinical severity of the regurgitation and the tempo of the illness as dictated by serial hemodynamic measurements, determine the rapidity with which one proceeds with emergent valve surgery. In the case of papillary muscle rupture, which is the cause of death in 1% to 5% of fatal myocardial infarctions, urgent surgical intervention is mandatory.45
Related posts:
 Evolution of the Coronary Care Unit: Past, Present, and Future
Evolution of the Coronary Care Unit: Past, Present, and Future
 Elevated Cardiac Troponin in the Absence of Acute Coronary Syndromes: Mechanism, Significance, and Prognosis
Elevated Cardiac Troponin in the Absence of Acute Coronary Syndromes: Mechanism, Significance, and Prognosis
 Conduction Disturbances in Acute Myocardial Infarction
Conduction Disturbances in Acute Myocardial Infarction
 Pharmacologic Interactions in the CICU
Pharmacologic Interactions in the CICU
 Acute Aortic Syndromes: Diagnosis and Management
Acute Aortic Syndromes: Diagnosis and Management
 Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


