Senior Dissertation
Univ. PA School of Medicine
1803
This chapter describes three types of movement disorder that you are likely to encounter in the ICU: (a) involuntary movements (i.e., seizures), (b) weak or ineffective movements (i.e., neuromuscular weakness), and (c) no movements (i.e., drug-induced paralysis). These disorders share one common trait, which is the ability to “waste” the human machine.
SEIZURES
Seizures are second only to metabolic encephalopathy as the most common neurological complication in critically ill patients (1). The incidence of new-onset seizures in ICU patients is 0.8–3.5% (1,2).
Types Of Seizures
Seizures are classified by the extent of brain involvement (generalized vs. focal seizures), the presence or absence of abnormal movements (convulsive vs. nonconvulsive seizures), and the type of movement abnormality (e.g., tonic-clonic, myoclonic movements).
Abnormal Movements
The movements associated with seizures can be tonic (caused by sustained muscle contraction), clonic (rhythmic movements with a regular amplitude and frequency), or myoclonic (irregular movements that vary in amplitude and frequency) (3). Some movements are familiar (e.g., chewing) but repetitive; these are called automatisms.
Generalized Seizures
Generalized seizures arise from synchronous, rhythmic electrical discharges that involve most of the cerebral cortex, and they are always associated with loss of consciousness (3). These seizures typically produce tonic-clonic movements of the extremities, but they can also occur without abnormal movements (generalized nonconvulsive seizures) (4).
Partial Seizures
Partial seizures can arise from diffuse or localized rhythmic discharges in the brain, and the clinical manifestations can vary widely, as demonstrated by the following two examples of partial seizures.
1. Partial complex seizures are nonconvulsive seizures that produce behavioral changes. The typical manifestation is a patient who is awake but not aware of the surroundings (similar to absence seizures). They are often preceded by an aura (e.g., a particular smell), and can be accompanied by repetitive chewing motions or lip smacking (automatisms).
2. Epilepsia partialis continua is a convulsive seizure that is characterized by persistent tonic-clonic movements of the facial and limb muscles on one side of the body.
Myoclonus
Myoclonus is characterized by irregular jerking movements of the extremities, which can occur spontaneously, or in response to painful stimuli or loud noises (startle myoclonus). These movements can be seen in any type of encephalopathy (metabolic, ischemic). In patients who do not awaken in the hours following a cardiac arrest, the presence of myoclonus that lasts longer than 24 hours carries a poor prognosis for neurologic recovery (5). Myoclonus is not universally regarded as a seizure, because it is not associated with rhythmic discharges on the EEG (6).
Status Epilepticus
Status epilepticus is traditionally defined as more than 30 minutes of either continuous seizure activity, or recurrent seizure activity without a period of recovery (6). Since generalized convulsive seizures are unlikely to stop after 5 minutes, a recently proposed definition of status epilepticus is 5 minutes of continuous seizure activity, or two seizures without an intervening period of consciousness (7). Status epilepticus can involve any type of seizure, and can be “convulsive” (i.e., associated with abnormal body movements) or “nonconvulsive” (i.e., not associated with abnormal body movements).
NONCONVULSIVE STATUS EPILEPTICUS: Most cases of nonconvulsive status epilepticus involve partial complex seizures (which are not common in ICU patients), but as many as 25% of generalized seizures can be nonconvulsive (8). Generalized nonconvulsive status epilepticus is also known as subtle status epilepticus, and typically occurs when generalized convulsive seizures have not been adequately treated (4). These seizures are associated with loss of consciousness, and they are a source of unexplained coma in the ICU. In one study, generalized nonconvulsive seizures were responsible for 8% of cases of coma in ICU patients (9). The diagnosis requires evidence of epileptiform discharges on the EEG.
Predisposing Conditions
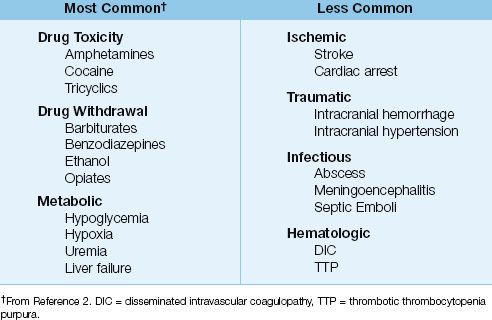
A variety of conditions can produce seizures in critically ill patients, as indicated in Table 45.1. In one survey of new-onset seizures in ICUs, the most common predisposing conditions were drug intoxication, drug withdrawal, and metabolic abnormalities (e.g., hypoglycemia) (2).
Table 45.1 Conditions That Promote Seizures in the ICU
Acute Management
The acute management described here pertains to generalized status epilepticus (GSE), both convulsive and nonconvulsive. The approach is divided into three stages, and the recommended drug dosing regimens for each stage are shown in Tables 45.2 and 45.3 (6,7).
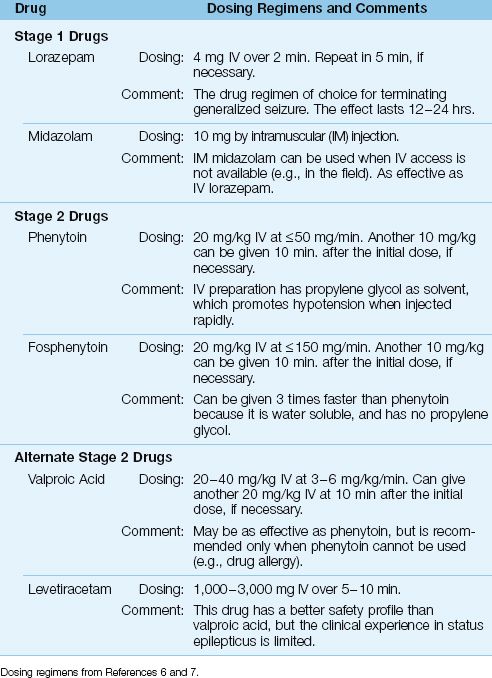
Stage 1 Drugs
The most effective drugs for rapid termination of generalized seizures are the benzodiazepines, which terminate 65–80% of convulsive seizures within 2–3 minutes (10,11).
LORAZEPAM: Intravenous lorazepam (4 mg IV over 2 minutes) is the drug regimen of choice for terminating GSE. The onset of action is less than two minutes, and the effect lasts for 12–24 hours (11,12).
MIDAZOLAM: The benefit of midazolam is rapid uptake when given by intramuscular (IM) injection. When intravenous access is not available or difficult to establish, midazolam can be given IM in a dose of 10 mg, and the efficacy in terminating GSE is equivalent to IV lorazepam (13). If the time to establish intravenous access is considered, IM midazolam produces more rapid suppression of seizures (3–4 minutes) than IV lorazepam (13). This approach is well suited for prehospital control of GSE.
Stage 2 Drugs
Stage 2 drugs are used for seizures that are refractory to benzodiaze-pines, or are likely to recur within 24 hours. The drug-of-choice for this purpose is phenytoin.
PHENYTOIN: The initial IV dose of phenytoin is 20 mg/kg, with a second dose of 10 mg/kg, if necessary. The goal is a serum phenytoin level of 10–20 ∝g/mL. Phenytoin cannot be infused at a rate above 50 mg/min because of the risk of cardiac depression and hypotension. This means that, for a 70 kg adult, the initial dose of phenytoin (20 mg/kg) will require 30 minutes to complete, and this is a disadvantage when GSE has not resolved (i.e., is refractory to benzodiazepines). The cardiac depression is attributed propylene glycol, which is used as a solvent in IV phenytoin preparations.
FOSPHENYTOIN: Fosphenytoin is a water-soluble phenytoin analogue that produces less cardiac depression than phenytoin because it does not contain propylene glycol. As a result, fosphenytoin can be infused three times faster than phenytoin (150 mg/min vs. 50 mg/min) (14). Fosphenytoin is a prodrug (must be converted to phenytoin), and is given in the same doses as phenytoin.
ALTERNATIVE DRUGS: Intravenous valproic acid (20–40 mg/kg) is as effective as phenytoin in terminating GSE (15), but is recommended only when phenytoin cannot be given (e.g., because of a drug allergy) (6,7). Another alternative to phenytoin is levetiracetam (1,000–3,000 mg IV), which has a better safety profile than valproic acid, but has not been evaluated as extensively.
Stage 3: Refractory Status Epilepticus
Ten percent of patients with GSE are refractory to stage 1 and 2 drugs (8). The recommended treatment at this point is anesthetic doses of one of the drugs in Table 45.3. Pentobarbital may be the favored drug in this situation (16). At this stage, a neurology consult is the best option.
Table 45.2 Drug Regimens for Generalized Status Epilepticus
Outcomes
The in-hospital mortality rates are as high as 21% for convulsive GSE, as high as 52% for nonconvulsive GSE, and as high as 61% for refractory GSE (7).
Table 45.3 Drug Regimens for Refractory Status Epilepticus

NEUROMUSCULAR WEAKNESS SYNDROMES
The following is a description of the acute neuromuscular weakness syndromes that you may encounter in an ICU.
Myasthenia Gravis
Myasthenia gravis (MG) is an autoimmune disease produced by antibody-mediated destruction of acetylcholine receptors on the postsynaptic side of neuromuscular junctions (17).
Predisposing Conditions
MG can be triggered by major surgery or a concurrent illness. Thymic tumors are responsible for up to 20% of cases, and hyperthyroidism is the culprit in 5% of cases. Several drugs can precipitate or aggravate MG (17). The principal offenders are antibiotics (e.g., aminoglycosides, ciprofloxacin) and cardiac drugs (e.g., beta-adrenergic blockers, lidocaine, procainamide, quinidine).
Clinical Features
The muscle weakness in MG has the following features:
1. The weakness worsens with activity and improves with rest.
2. Weakness is first apparent in the eyelids and extraocular muscles, and limb weakness follows in 85% of cases (19).
3. Progressive weakness often involves the chest wall and diaphragm, and rapid progression to respiratory failure, called myasthenic crisis, occurs in 15–20% of patients (18).
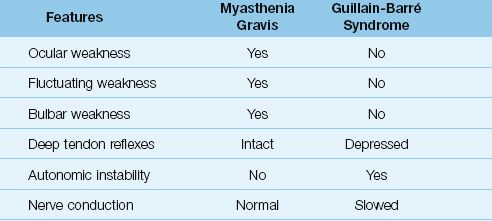
4. The deficit is purely motor, and deep tendon reflexes are preserved (see Table 45.4).
Diagnosis
The diagnosis of MG is suggested by weakness in the eyelids or extraocular muscles that worsens with repeated use. The diagnosis is confirmed by: (a) increased muscle strength after the administration of edrophonium (Tensilon), an acetylcholinesterase inhibitor, and (b) a positive assay for acetylcholine receptor antibodies in the blood, which are present in 85% of patients of MG (17).
Treatment
The first line of therapy is an acetylcholinesterase inhibitor, pyridostigmine (Mestinon), which is started at an oral dose of 60 mg every 6 hours, and can be increased to 120 mg every 6 hours if necessary (20,21). Pyridostigmine can be given intravenously to treat myasthenic crisis: the IV dose is 1/30th of the oral dose (19,20).
Immunotherapy is added, if needed, using either prednisone (1–1.5 mg/kg/day), azathioprine (1–3 mg/kg/day), or cyclosporine (2.5 mg/kg twice daily) (21). To reduce the need for long-term immunosuppressive therapy, surgical thymectomy is often advised in patients under 60 years of age (21).
ADVANCED CASES: In advanced cases requiring mechanical ventilation, there are two treatment options: (a) plasmapharesis to clear pathological antibodies from the bloodstream, or (b) administration of immunoglobulin G (0.4 or 2 gm/kg/day IV for 2–5 days) to neutralize the pathologic antibodies. Both approaches are equally effective (19,21), but plasma-pharesis produces a more rapid response (21).
Table 45.4 Comparative Features of Myasthenia Gravis and Guillain-Barré Syndrome
Guillain-Barré Syndrome
The Guillain-Barré syndrome is a subacute inflammatory demyelinating polyneuropathy that often follows an acute infectious illness (by 1–3 weeks) (22,23). An immune etiology is suspected.
Clinical Features
The presenting features of the Guillain-Barré syndrome include paresthesias and symmetric limb weakness that evolves over a period of a few days to a few weeks. Progression to respiratory failure occurs in 25% of cases (22), and autonomic instability can be a feature in advanced cases (24). The condition resolves spontaneously in about 80% of cases, but residual neurological deficits are common (22).
Diagnosis
The diagnosis of Guillain-Barré syndrome is based on the clinical presentation (paresthesias and symmetric limb weakness), nerve conduction studies (slowed conduction), and cerebrospinal fluid analysis (elevated protein in 80% of cases) (22). The features that distinguish the Guillain-Barré syndrome from myasthenia gravis are shown in Table 45.4.
Treatment
Treatment is mostly supportive, but in advanced cases with respiratory failure, plasmapharesis or intravenous immunoglobulin G (0.4 g/kg/day for 5 days) are equally effective in producing short-term improvement (23). Immunoglobulin G is often preferred because it is easier to carry out.
Critical Illness Neuromyopathy
The disorders known as critical illness polyneuropathy (CIP) and critical illness myopathy (CIM) are secondary disorders, and typically accompany severe sepsis and other conditions associated with progressive systemic inflammation (25). These disorders often co-exist in the same patient, and become apparent when patients fail to wean from mechanical ventilation.
Pathogenesis
CIP is a diffuse sensory and motor axonal neuropathy that is discovered in at least 50% of patients with severe sepsis and septic shock (25–27). The onset is variable, occurring from 2 days to a few weeks after the onset of the septic episode. CIP is considered the most common peripheral neuropathy in critically ill patients (28)
CIM is a diffuse inflammatory myopathy that involves both limb and truncal muscles (29). Predisposing conditions include severe sepsis and septic shock, and prolonged periods of drug-induced neuromuscular paralysis, particularly when combined with high-dose corticosteroid therapy (25,26,29). CIM has also been reported in one-third of patients with status asthmaticus who are treated with high-dose corticosteroids (29).
Clinical Features
As just mentioned, CIP and CIM often go undetected until there is an unexplained failure to remove a patient from mechanical ventilation. Physical examination will then reveal a flaccid quadriparesis with hypo-reflexia or areflexia. The diagnosis of CIP can be confirmed by nerve conduction studies (which show slowed conduction in sensory and motor fibers) (27), and the diagnosis of CIM can be confirmed by electromyography (which shows myopathic changes) and by muscle biopsy (which shows atrophy, loss of myosin filaments, and inflammatory infiltration) (29).
There is no treatment for CIP or CIM. Complete recovery is expected in about half the patients (27), but it can takes months to recover.
DRUG-INDUCED PARALYSIS
Drug-induced paralysis is used in the following situations: (a) to facilitate endotracheal intubation, (b) to prevent shivering during induced hypothermia (in comatose survivors of cardiac arrest), and (c) to facilitate mechanical ventilation in severely agitated patients (30). The latter practice is frowned upon for reasons stated later.
Neuromuscular blocking agents act by binding to acetylcholine receptors on the postsynaptic side of the neuromuscular junction. Once bound, there are two different modes of action: (a) depolarizing agents act like acetylcholine and produce a sustained depolarization of the post-synaptic membrane, and (b) non-depolarizing agents act by inhibiting depolarization of the post-synaptic membrane.
Commonly Used Agents
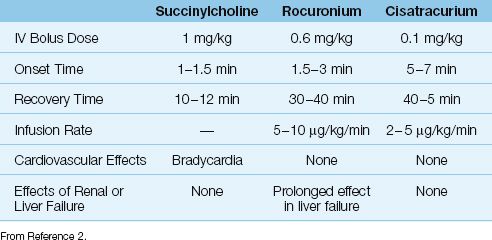
The comparative features of three commonly used neuromuscular blocking agents are shown in Table 45.5 (31).
Table 45.5 Features of Commonly Used Neuromuscular Blocking Agents
Succinylcholine
Succinylcholine is a depolarizing agent with a rapid onset of action (60–90 seconds) and a rapid recovery time (10–12 minutes). Because of these features, succinylcholine is used to facilitate endotracheal intubation.
SIDE EFFECTS: Succinylcholine-induced depolarization of skeletal muscle promotes the efflux of potassium from muscle cells. This can be associated with a 0.5 mEq/L rise in the serum potassium (32), but this effect is transient and without consequence. However, life-threatening hyperkalemia can occur when succinylcholine is given to patients with skeletal muscle “denervation injury” (e.g., from head or spinal cord injury), or patients with rhabdomyolysis, burns, or chronic immobility. As a result, succinylcholine is not advised for patients with these conditions.
Rocuronium
Rocuronium is a non-depolarizing neuromuscular blocker with a rapid onset of action (1.5–3 minutes) and an “intermediate” recovery time (30–40 min). Because of the rapid onset of action, rocuronium can be used for endotracheal intubation when succinylcholine is not advised. However, larger doses (1 mg/kg) are required for intubation, and this will prolong the recovery time (31). Rocuronium can be infused at a rate of 5–10 ∝g/kg/min for prolonged neuromuscular paralysis. The drug is well tolerated, and has no cardiovascular side effects. Rocuronium has gradually replaced a related neuromuscular blocker, vecuronium, be-cause of its rapid onset of action.
Cisatracurium
Cisatracurium is a non-depolarizing agent with a prolonged onset of action (5–7 min) and an “intermediate” recovery time. It is an isomer of atracurium (another neuromuscular blocker), and was developed to eliminate the histamine release associated with atracurium. Cisatracur-ium can be infused at a rate of 2–5 ∝g/kg/min for prolonged neuromuscular paralysis, and the drug is well suited for ICU patients because blood levels are not influenced by renal or liver dysfunction (31).
Monitoring
The standard method of monitoring drug-induced paralysis is to apply a series of four low-frequency (2 Hz) electrical pulses to the ulnar nerve at the forearm, and observe for adduction of the thumb. Total absence of thumb adduction is evidence of excessive block. The desired goal is 1 or 2 perceptible twitches, and the drug infusion is adjusted to achieve that end-point (30).
Avoiding Drug-Induced Paralysis
The experience of being awake while paralyzed is both horrifying and painful (33), and it is imperative to keep patients heavily sedated while they are paralyzed. However, it is not possible to evaluate the adequacy of sedation or pain control while a patient is paralyzed. The inability to insure adequate sedation and pain control is the major reason to avoid drug-induced paralysis whenever possible. Avoiding prolonged periods of neuromuscular paralysis will also reduce the risk of the following complications:
1. Critical illness myopathy (explained earlier).
2. “Hypostatic” pneumonia (from pooling of respiratory secretions in dependent lung regions).
3. Venous thromboembolism (from prolonged immobilization).
4. Pressure ulcers on the skin (also from prolonged immobilization).
A FINAL WORD
Inflammation Strikes Again
One of the central themes in this book is the widespread harm inflicted by progressive systemic inflammation in critically ill patients. Inflam-matory injury is responsible for the acute respiratory distress syndrome (see Chapter 23) and acute kidney injury (see Chapter 34), as well as the multiorgan failure associated with septic shock (see Chapter 14). In this chapter, we learn that inflammation can also damage peripheral nerves (i.e., critical illness polyneuropathy) and skeletal muscle (i.e., critical illness myopathy). These injuries support the notion that inflammation is the most lethal force that you will face in the ICU.
REFERENCES
Seizures
1. Bleck TP, Smith MC, Pierre-Louis SJ, et al. Neurologic complications of critical medical illnesses. Crit Care Med 1993; 21:98–103.
2. Wijdicks EF, Sharbrough FW. New-onset seizures in critically ill patients. Neurology 1993; 43:1042–1044.
3. Chabolla DR. Characteristics of the epilepsies. Mayo Clin Proc 2002; 77:981–990.
4. Holtkamp M, Meierkord H. Nonconvulsive status epilepticus: a diagnostic and therapeutic challenge in the intensive care setting. Ther Adv Neurol Disorders 2011; 4:169–181.
5. Wijdicks EF, Parisi JE, Sharbrough FW. Prognostic value of myoclonus status in comatose survivors of cardiac arrest. Ann Neurol 1994; 35:239–243.
6. Meierkord H, Boon P, Engelsen B, et al. EFNS guideline on the management of status epilepticus in adults. Eur J Neurol 2010; 17:348–355.
7. Brophy GM, Bell R, Claassen J, et al. Guidelines for the evaluation and management of status epilepticus. Neurocrit Care 2012; 17:3–23.
8. Marik PE, Varon J. The management of status epilepticus. Chest 2004; 126:582–591.
9. Towne AR, Waterhouse EJ, Boggs JG, et al. Prevalence of nonconvulsive status epilepticus in comatose patients. Neurology 2000; 54:340–345.
10. Treiman DM, Meyers PD, Walton NY, et al. A comparison of four treatments for generalized convulsive status epilepticus. N Engl J Med 1998; 339:792–798.
11. Lowenstein DH, Alldredge BK. Status epilepticus. N Engl J Med 1998; 338:970–976.
12. Manno EM. New management strategies in the treatment of status epilepticus. Mayo Clin Proc 2003 ;78:508–518.
13. Silbergleit R, Durkalsi V, Lowenstein D, et al. Intramuscular versus intravenous therapy for prehospital status epilepticus. N Engl J Med 2012; 366:591–600.
14. Fischer JH, Patel TV, Fischer PA. Fosphenytoin: clinical pharmacokinetics and comparative advantages in the acute treatment of seizures. Clin Pharmacokinet 2003; 42:33–58.
15. Misra UK, Kalita J, Patel R. Sodium valproate vs. phenytoin in status epilepticus: a pilot study. Neurology 2006; 67:340–342.
16. Claassen J, Hirsch LJ, Emerson RG, et al. Treatment of refractory status epilepticus with pentobarbital, propofol, or midazolam: a systematic review. Epilepsia 2002; 43:146–153.
Myasthenia Gravis
17. Vincent A, Palace J, Hilton-Jones D. Myasthenia gravis. Lancet 2001; 357:2122–2128.
18. Wittbrodt ET. Drugs and myasthenia gravis. An update. Arch Intern Med 1997; 157:399–408.
19. Drachman DB. Myasthenia gravis. N Engl J Med 1994; 330:1797–1810.
20. Berrouschot J, Baumann I, Kalischewski P, et al. Therapy of myasthenic crisis. Crit Care Med 1997; 25:1228–1235.
21. Saperstein DS, Barohn RJ. Management of myasthenia gravis. Semin Neurol 2004; 24:41–48.
Guillain-Barré Syndrome
22. Hughes RA, Cornblath DR. Guillain-Barré syndrome. Lancet 2005; 366:1653–1666.
23. Hund EF, Borel CO, Cornblath DR, et al. Intensive management and treatment of severe Guillain-Barré syndrome. Crit Care Med 1993; 21:433–446.
24. Pfeiffer G, Schiller B, Kruse J, et al. Indicators of dysautonomia in severe Guillain-Barré syndrome. J Neurol 1999; 246:1015–1022.
Critical Illness Neuromyopathy
25. Hund E. Neurological complications of sepsis: critical illness polyneuropathy and myopathy. J Neurol 2001; 248:929–934.
26. Bolton CF. Neuromuscular manifestations of critical illness. Muscle & Nerve 2005; 32:140–163.
27. van Mook WN, Hulsewe-Evers RP. Critical illness polyneuropathy. Curr Opin Crit Care 2002; 8:302–310.
28. Maramatton BV, Wijdicks EFM. Acute neuromuscular weakness in the intensive care unit. Crit Care Med 2006; 34:2835–2841.
29. Lacomis D. Critical illness myopathy. Curr Rheumatol Rep 2002; 4:403–408.
Drug-Induced Paralysis
30. Murray MJ, Cowen J, DeBlock H, et al. Clinical practice guidelines for sustained neuromuscular blockade in the adult critically ill patient. Crit Care Med 2002; 30:142–156.
31. Donati F, Bevan DR. Neuromuscular blocking agents. In: Barash PG, Cullen BF, Stoelting RK, et al, eds. Clinical Anesthesia. 6th ed. Philadelphia: Lippincott Williams & Wilkins, 2009:498–530.
32. Koide M, Waud BE. Serum potassium concentrations after succinylcholine in patients with renal failure. Anesthesiology 1972; 36:142–145.
33. Parker MM, Schubert W, Shelhamer JH, et al. Perceptions of a critically ill patient experiencing therapeutic paralysis in an ICU. Crit Care Med 1984; 12:69–71.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


