1875
The earliest sea-living organisms showed a preference for intracellular potassium and a disdain for intracellular sodium, and this behavior eventually changed the composition of the oceans from a potassium salt solution to a sodium salt solution. The same preferences are found in the human organism, where 98% of the total body potassium is located inside cells, and only 2% remains in the extracellular fluid (1–3). As a result, monitoring the plasma (extracellular) potassium level as an index of total body potassium is like evaluating the size of an iceberg by its tip. With this limitation in mind, this chapter describes the causes and consequences of abnormalities in the plasma potassium concentration.
BASICS
Potassium Distribution
The intracellular preponderance of potassium is the result of a sodium-potassium (Na+– K+) exchange pump on cell membranes that moves Na+ out of cells and moves K+ into cells in a 3:2 ratio (1). One of the major roles of this pump is to create a voltage gradient across cell membranes in ”excitable” tissues (i.e., nerves and muscle), which allows for the transmission of electrical impulses in these tissues.
The small fraction of total body K+ that is present outside cells is illustrated in Figure 36.1. The total body potassium in healthy adults is about 50–55 mEq per kg body weight (1). Using the conservative estimate of 50 mEq/kg in a 70 kg adult yields a total body potassium of 3,500 mEq, with 70 mEq (2%) in extracellular fluid. Because the plasma accounts for about 20% of the extracellular fluid, the potassium content of plasma will be about 15 mEq, which is only 0.4% of the total body potassium. This emphasizes the limited size of the K+ pool that is available for evaluating total body potassium.
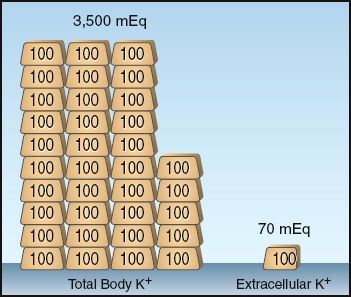
FIGURE 36.1 Illustration of the small fraction of total body K+ that is present outside cells. This example pertains to a 70 kg adult with an estimated total body K+ of 50 mEq/kg body weight. Each gold bar represents 100 mEq of K+.
Serum Potassium
The relationship between total body K+ and serum (plasma) K+ is shown in Figure 36.2 (4,5). Note the curvilinear shape of the curve, with the flat portion of the curve in the region of potassium deficiency. In an averaged-size adult with a normal serum K+ of 4 mEq/L, a total body K+ deficit of 200–400 mEq is required to produce a decrease in plasma K+ of 1 mEq/L, while a total body K+ excess of 100–200 mEq is required to produce a similar (1 mEq/L) increase in plasma K (5). Therefore, for a given change in serum K+, the change in total body K+ is twofold greater with K+ depletion (hypokalemia) than with K+ excess (hyperkalemia). The larger deficit associated with hypokalemia is due to the large pool of intracellular K+ that can replenish extracellular K+ (and help to maintain serum K+) when K+ is lost.
Potassium Excretion
Small amounts of K+ are lost in stool (5–10 mEq/day) and sweat (0–10 mEq/day), but the majority of K+ loss is in urine (40–120 mEq/day, depending on K+ intake) (1).
Renal Excretion
Most of the K+ that is filtered at the glomerulus is passively reabsorbed in the proximal tubules (along with sodium and water), and K+ is then secreted in the distal tubules and collecting ducts (1). Potassium excretion in urine is primarily a function of K+ secretion in the distal nephron, which is controlled by plasma K+ and (primarily by ) aldosterone. When renal function is normal, the capacity for renal K+ excretion is great enough to prevent a sustained rise in serum K+ in response to an increased K+ load (1).
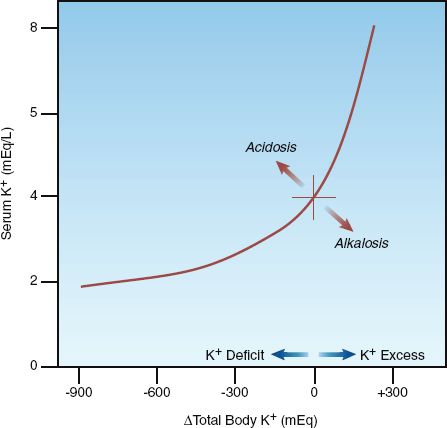
FIGURE 36.2 Relationship between the serum potassium concentration and total body potassium. Redrawn from Reference 4.
ALDOSTERONE: Aldosterone is a mineralocorticoid that is released by the adrenal cortex in response to an increase in plasma K+ (and angiotensin II), and it increases K+ excretion in the urine by stimulating K+ secretion in the distal nephron. Potassium secretion is linked to sodium reabsorption, so aldosterone also promotes sodium and water retention. The diuretic spironolactone acts by blocking the actions of aldosterone in the kidneys. As a result, spironolactone is a potassium-sparing diuretic.
HYPOKALEMIA
Hypokalemia (serum K+ <3.5 mEq/L) can be the result of K+ movement into cells (transcellular shift), or a decrease in total body K+ ( K+ depletion) (3–6).
Transcellular Shift
The movement of K+ into cells is facilitated by stimulation of β2 adrenergic receptors on cell membranes in muscle, and this explains the decrease in serum K+ associated with inhaled β2-agonist bronchodilators (e.g., albuterol) (7). This effect is mild (″0.5 mEq/L) in the usual therapeutic doses (7), but is more significant when inhaled β2-agonists are used in combination with diuretics (8). Other conditions that promote K+ movement into cells include alkalosis (respiratory or metabolic), hypothermia (accidental or induced), and insulin. Alkalosis has a variable and unpredictable effect on serum K+ (9). Hypothermia causes a transient drop in serum K+ that resolves on rewarming (10).
Potassium Depletion
Potassium depletion can be the result of K+ loss via the kidneys or gastrointestinal tract. The site of K+ loss (renal or extrarenal) is usually obvious, but can be identified by measuring the urinary K+ and chloride concentrations, as shown in Figure 36.3.
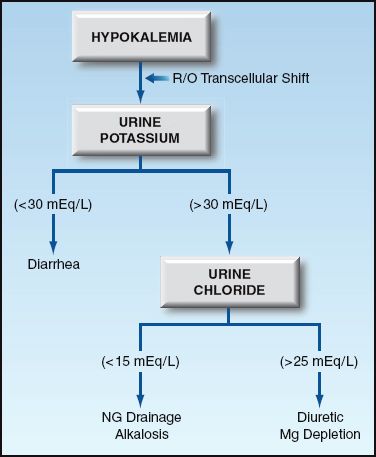
FIGURE 36.3 Diagnostic approach to hypokalemia.
Renal Potassium Loss
The leading cause of renal K+ loss is diuretic therapy. Other causes likely to be seen in the ICU include nasogastric drainage, alkalosis, and magnesium depletion. Nasogastric drainage has a low concentration of K+ (10–15 mEq/L), but the resulting loss of volume and H+ promotes K+ loss in the urine. The urine chloride is low (<15 mEq/L) with nasogastric drainage and alkalosis, and it is high (>25 mEq/L) with diuretic therapy and magnesium depletion. Magnesium depletion impairs K+ reabsorption in the renal tubules and may play a very important role in promoting K+ depletion in critically ill patients, particularly those receiving diuretics (12).
Extrarenal Potassium Loss
The major cause of extrarenal K+ loss is diarrhea. Normal K+ loss in stool is only 5–10 mEq daily. In secretory and inflammatory diarrhea, the concentration of K+ in stool is 15–40 mEq/L, and the daily stool volume can reach 10 liters in severe cases. Therefore, K+ losses can reach 400 mEq daily in severe cases of inflammatory or secretory diarrhea (11).
Clinical Manifestations
Severe hypokalemia (serum K+ <2.5 mEq/L) can be associated with diffuse muscle weakness (3), but in most cases, hypokalemia is asymptomatic. Abnormalities in the ECG are the major manifestation of hypokalemia, and can be present in 50% of cases (13). The ECG abnormalities include prominent U waves (more than 1 mm in height), flattening and inversion of T waves, and prolongation of the QT interval. However, these changes are not specific for hypokalemia; i.e., the T wave changes and U waves can be seen with digitalis or left ventricular hypertrophy, and QT prolongation can be the result of drugs, hypocalcemia, or hypomagnesemia.
Arrhythmias
Contrary to popular belief, hypokalemia alone is not a risk for serious arrhythmias (3,13). However, hypokalemia can add to the risk of serious arrhythmias from other conditions (e.g., myocardial ischemia) (3).
Management of Hypokalemia
The first concern in hypokalemia is to eliminate or treat any condition that promotes transcellular potassium shifts (e.g., alkalosis) (3). If the hypokalemia is due to K+ depletion, proceed as described next.
Estimate Potassium Deficits
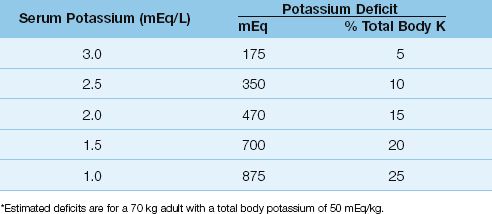
About 10% of total body K+ is lost for every 1 mEq/L decrease in serum K+ (14). For a 70 kg adult with a normal total body K+ of 50 mEq/kg, the estimated K+ deficits associated with progressive hypokalemia are shown in Table 36.1. Note that even mild hypokalemia (serum K+ = 3 mEq/L) is associated with a considerable K+ deficit (175 mEq).
Table 36.1 Potassium Deficits in Hypokalemia*
Potassium Replacement
FLUIDS: The usual replacement fluid is potassium chloride, which is available as a concentrated solution (1–2 mEq/mL) in ampules containing 10, 20, 30, and 40 mEq of potassium. These solutions are extremely hyperosmotic (the 2 mEq/mL solution has an osmolality of 4,000 mosm/kg H2O) and must be diluted (15). A potassium phosphate solution is also available that contains 4.5 mEq potassium and 3 mmol phosphate per mL, and this solution is preferred by some for potassium replacement in diabetic ketoacidosis (because of the phosphate depletion in diabetic ketoacidosis).
RATE OF REPLACEMENT: The standard method of intravenous K+ replacement is to add 20 mEq of K+ to 100 mL of isotonic saline and infuse this mixture over 1 hour (16). The maximum rate of intravenous potassium replacement is usually set at 20 mEq/hr (16), but dose rates of 40 mEq/hour may be necessary (e.g., serum K+ <1.5 mEq/L or serious arrhythmias), and dose rates as high as 100 mEq/hour have been used safely (17). Infusion through a large, central vein is preferred, if possible, because of the irritating properties of the hyperosmotic KCL solutions. However, delivery into the superior vena cava is not recommended if the desired rate of replacement exceeds 20 mEq/hr because there is a (poorly documented) risk of an abrupt rise in plasma K+ in the right heart chambers severe enough to produce asystole.
RESPONSE: The serum K+ may be slow to rise initially, as predicted by the flat portion of the curve in Figure 36.2. If the hypokalemia is resistant or refractory to K+ replacement, magnesium depletion should be considered. Magnesium depletion promotes urinary K+ loss (as described earlier), and in patients who are magnesium deficient, hypokalemia is often refractory to K+ replacement until the magnesium is repleted (18). Magnesium deficiency may play an important role in diuretic-induced hypokalemia, as described in the next chapter.
HYPERKALEMIA
While hypokalemia is often well tolerated, hyperkalemia (serum K+ >5.5 mEq/L) can be a life-threatening condition (2,3,19,20).
Etiology
Hyperkalemia can be the result of potassium release from cells (transcellular shift), or impaired renal excretion of potassium. If the source of the hyperkalemia is unclear, a spot urine K+ can be useful. A high urine K+ (>30 mEq/L) suggests a transcellular shift, and a low urine K+ (<30 mEq/L) indicates impaired renal excretion. If the hyperkalemia is unexpected, the condition described next should be considered.
Pseudohyperkalemia
Hyperkalemia that is present ex vivo (in the blood sample), but not in vivo, is known as pseudohyperkalemia. The leading cause of this condition is potassium release from traumatic hemolysis during the venipuncture. This is more common than suspected, and has been reported in 20% of blood samples with hyperkalemia (21). Other sources of pseudohyperkalemia in-clude: (a) K+ release from muscles during fist clenching (22), and (b) potassium release from clot formation in the blood collection tube in patients with severe leukocytosis (>50,000/mm3) or severe thrombocytosis (platelet count >1 million/mm3). When pseudohyperkalemia is suspected, a repeat blood sample should be obtained using precautions to mitigate the suspected problem (e.g., minimize suction when withdrawing blood).
Transcellular Shift
The conditions associated with K+ movement out of cells include acidosis, rhabdomyolysis, tumor lysis syndrome, drugs, and blood transfusions.
ACIDOSIS: The presumed mechanism for the relationship between acidosis and hyperkalemia is competition between H+ and K+ for the same site on the membrane pump that moves K+ into cells. However, a causal link between acidosis and hyperkalemia is being questioned because the organic acidoses (lactic acidosis and ketoacidosis) are not associated with hyperkalemia (9), and respiratory acidosis has an inconsistent and unpredictable association with hyperkalemia (9).
TUMOR LYSIS SYNDROME: Tumor lysis syndrome is an acute, life-threatening condition that appears within 7 days after the initiation of cytotoxic therapy for selected malignancies (e.g., non-Hodgkins lymphoma). Characteris-tic features include the combination of hyperkalemia, hyperphosphatemia, hypocalcemia, and hyperuricemia, often accompanied by acute kidney injury (23). Hyperkalemia is the most immediate threat to life.
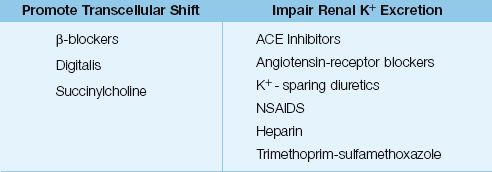
DRUGS: The drugs that promote K+ movement out of cells are listed in Table 36.2. Digitalis inhibits the membrane Na+– K+ exchange pump, but hyperkalemia occurs only with acute (not chronic) digitalis toxicity (24). Succinylcholine is an ultra short-acting neuromuscular blocking agent that also inhibits the membrane Na+– K+ exchange pump (a depolarizing effect), and this effect is associated with a minor increase in serum K+ (<1 mEq/L) that lasts only 5–10 minutes (25). Life-threatening increases in serum K+ have been reported when succinylcholine is used in patients with “denervation injury”of skeletal muscle (e.g., spinal cord injury). This is attributed to a an exaggerated response to depolarizing signals following denervation (denervation hypsersensitivity).
Table 36.2 Drugs That Promote Hyperkalemia in the ICU
Impaired Renal Excretion
As mentioned earlier in the chapter, when renal function is normal, the capacity for urinary K+ excretion is great enough to prevent a sustained increase in serum K+ in response to a K+ load (1). As a result, hyperkalemia always involves a defect in renal K+ excretion. The common causes of impaired renal K+ excretion include renal failure, adrenal insufficiency, and drugs. In renal failure, hyperkalemia usually doesn’t occur until the GFR drops below 10 mL/min, but it can appear earlier when renal failure is the result of interstitial nephritis. Adrenal insufficiency impairs renal potassium excretion, but hyperkalemia is seen only in chronic adrenal insufficiency.
DRUGS: Drugs that impair renal potassium excretion represent a common source of hyperkalemia. A list of common offenders is shown in Table 36.2 (25–29). The drugs most often implicated are angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, potassium sparing diuretics, and nonsteroidal antiinflammatory drugs. All of these agents promote hyperkalemia by inhibiting the renin–angiotensin–aldosterone system. The hyperkalemia from these drugs often occurs in combination with K+ supplements or renal insufficiency.
Blood Transfusion
Hyperkalemia is a recognized (but inconsistent) complication of massive blood transfusion (i.e., blood replacement equivalent to the blood volume). The temperature used to store red blood cells (4ºC) shuts off the Na+– K+ exchange pump in the erythrocyte cell membrane, and this results in a steady leakage of K+ from the cells (30). The K+ concentration in the supernatant increases steadily as the storage time increases. After 18 days of storage (the average time that blood is stored), the potassium load in one unit of packed red blood cells is 2 to 3 mEq (30), so massive transfusion (usually at least 6 units of red blood cells) represents a K+ load of at least 12–18 mEq. This is a considerable load, considering that plasma contains about 9–10 mEq of K+ in an average-sized adult.
The K+ load in transfused blood is normally cleared by the kidneys, but when systemic blood flow is compromised (which applies to most patients who need massive blood transfusions), renal K+ excretion is impaired, and the K+ infused in blood transfusions will accumulate. The transfusion volume needed to produce hyperkalemia will vary, but one study has shown that hyperkalemia begins to appear after transfusion of 7 units of red blood cells (31).
Cautopyreiophagia
In 1985, a case report was published describing a patient with severe hyperkalemia who was discovered to be ingesting 1,500 burnt match heads daily (a pica) (32). It turns out that burnt match heads are rich in potassium chlorate, and the habit of eating burnt match heads is known as cautopyreiophagia. This case is mentioned to demonstrate that anything is possible in medicine, and that there is a name for everything.
Clinical Consequences
The principal threat of hyperkalemia is slowed impulse transmission in the heart (from depolarization of cardiac muscle), which can progress to heart block and bradycardic cardiac arrest.
ECG Abnormalities
The ECG changes in progressive hyperkalemia are shown in Figure 36.4. The earliest change is the appearance of a tall, tapering (tented) T wave that is most evident in precordial leads V2 and V3. As the hyperkalemia progresses, the P wave amplitude decreases and the PR interval lengthens. The P waves eventually disappear and the QRS complex widens. The final event is ventricular fibrillation or asystole.
ECG changes usually begin to appear when the serum K+ reaches 7 mEq/L (33), but the threshold for ECG changes can vary widely. This is demonstrated by one case report that showed no ECG abnormalities at a serum K+ of 14 mEq/L (!) (34). Because of the variable relationship between the serum K+ and the ECG, both are used for management decisions in hyperkalemia.
Management of Severe Hyperkalemia
Severe hyperkalemia is defined as a serum K+ >6.5 mEq/L, or any serum K+ associated with ECG changes (33). The management of this condition has 3 goals: (a) antagonism of the cardiac effects of hyperkalemia, (b) transcellular shift of K+ into cells, and (c) removal of excess K+ from the body. The methods used to achieve these goals are described next, and are summarized in Table 36.3.
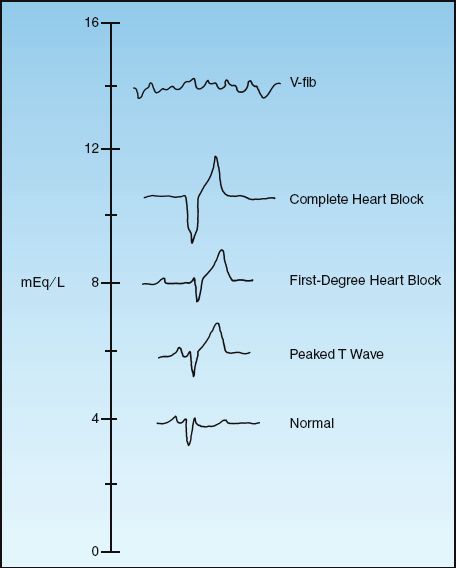
FIGURE 36.4 ECG abnormalities in progressive hyperkalemia.
Membrane Antagonism
Calcium increases the electrical charge difference across myocardial cell membranes and opposes the depolarization produced by hyperkalemia (35). The preferred calcium preparation is calcium gluconate, which is given in the dosing regimen shown in Table 36.3. The response to calcium is short-lived (20–30 minutes) and it does not reduce the serum K+, so other measures (e.g., insulin-glucose) should be initiated to reduce serum K+ levels. Calcium must be used cautiously in patients receiving digitalis because hypercalcemia aggravates digitalis cardiotoxicity. For patients receiving digitalis, calcium gluconate can be added to 100 mL of isotonic saline and infused over 20 to 30 minutes. Calcium is contraindicated if the hyperkalemia is a manifestation of digitalis toxicity.
Table 36.3 Management of Severe Hyperkalemia
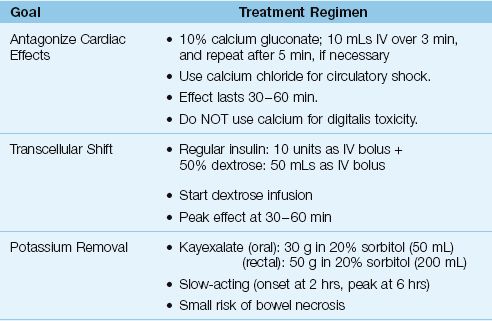
When hyperkalemia is associated with circulatory shock or cardiac arrest, calcium chloride is preferred to calcium gluconate. One ampule (10 mL) of 10% calcium chloride has three times the elemental calcium in one ampule of 10% calcium gluconate (270 mg vs. 90 mg, respectively), and the extra calcium has a potential advantage by promoting cardiac output and preserving peripheral vascular tone. The osmolality of calcium chloride is 2,000 mosm/kg H2O, so administration through a free-flowing central venous catheter is strongly advised.
Transcellular Shift
INSULIN-DEXTROSE: Insulin drives K+ into skeletal muscle cells by activating the membrane Na+– K+ exchange pump (36), and the insulin-dextrose regimen in Table 36.3 will decrease the serum K+ by at least 0.6 mEq/L (33). A dextrose infusion is advised after insulin-dextrose (unless the patient is hyperglycemic) because there is a risk of hypoglycemia at one hour (33). In the setting of hyperglycemia, insulin should be used without dextrose (33). The insulin effect is temporary (peak effect at 30–60 min), so measures that promote K+ removal should be started.
ß2-AGONISTS: The dose of inhaled β2– agonists (e.g., albuterol) needed to produce a significant (0.5–1 mEq/L) drop in serum K+ is at least 4 times the therapeutic dose (33), and this can produce unwanted side effects (e.g., tachycardia). Therefore, these agents are not advised for severe hyperkalemia.
BICARBONATE: There are two reasons to avoid bicarbonate for the management of severe hyperkalemia: (a) short-term infusions of bicarbonate (up to four hours) have no effect on serum K+ levels (33), and (b) bicarbonate can form complexes with calcium, which is counterproductive when calcium is used to antagonize the membrane effects of hyperkalemia.
Potassium Removal
Excess K+ can be removed via the bowel (with a cation-exchange resin) or directly from the bloodstream (with hemodialysis).
CATION EXCHANGE RESIN: Sodium polystyrene sulfonate (Kayexalate) is a cation exchange resin that promotes K+ clearance across the bowel mucosa. It can be given orally (preferred) or by retention enema, using the doses shown in Table 36.3. Kayexalate is usually mixed with sorbitol to prevent concretions. Each gram of resin binds 0.65 mEq of K+, and at least 6 hours is required for maximum effect (33). There are several case reports of necrotic lesions in the bowel linked to Kayexalate (37). Although this is an uncommon complication, the mortality rate is high (33%) (37).
HEMODIALYSIS: The most effective method of potassium removal is hemodialysis, which can produce a 1 mEq/L drop in serum K+ after one hour, and a 2 mEq/L drop after 3 hours (33).
A FINAL WORD
The following points in this chapter deserve emphasis.
1. Plasma K+ is only the “tip of the iceberg” in terms of evaluating total body potassium.
2. Potassium deficits are often larger than suspected in hypokalemia, even in mild cases (see Table 36.1).
3. Hypokalemia is remarkably well tolerated, and is not a risk for serious arrhythmias unless combined with another arrhythmogenic condition (e.g., acute coronary syndrome).
4. When hypokalemia is resistant or refractory to K+ replacement, magnesium deficiency is the likely culprit.
5. When using calcium to antagonize the cardiac effects of hyperkalemia, make sure to decrease the serum K+ (insulin-glucose is best), and begin measures to remove excess K+ (e.g., Kayexalate).
REFERENCES
Basics
1. Rose BD, Post TW. Potassium homeostasis. In: Clinical physiology of acid-base and electrolyte disorders. 5th ed. New York, NY: McGraw-Hill, 2001; 372–402.
2. Alfonzo AVM, Isles C, Geddes C, Deighan C. Potassium disorders—clinical spectrum and emergency management. Resusc 2006; 70:10–25.
3. Schaefer TJ, Wolford RW. Disorders of potassium. Emerg Med Clin North Am 2005; 23:723–747, viii–ix.
4. Brown RS. Extrarenal potassium homeostasis. Kidney Int 1986; 30:116–127.
5. Sterns RH, Cox M, Feig PU, et al. Internal potassium balance and the control of the plasma potassium concentration. Medicine 1981; 60:339–354.
Hypokalemia
6. Glover P. Hypokalaemia. Crit Care Resusc 1999; 1:239–251.
7. Allon M, Copkney C. Albuterol and insulin for treatment of hyperkalemia in hemodialysis patients. Kidney Int 1990; 38:869–872.
8. Lipworth BJ, McDevitt DG, Struthers AD. Prior treatment with diuretic augments the hypokalemic and electrocardiographic effects of inhaled albuterol. Am J Med 1989; 86:653–657.
9. Adrogue HJ, Madias NE. Changes in plasma potassium concentration during acute acid-base disturbances. Am J Med 1981; 71:456–467.
10. Bernard SA, Buist M. Induced hypothermia in critical care medicine: a review. Crit Care Med 2003; 31:2041–2051.
11. Gennari FJ, Weise WJ. Acid-base disturbances in gastrointestinal disease. Clin J Am Soc Nephrol 2008; 3:1861–1868.
12. Salem M, Munoz R, Chernow B. Hypomagnesemia in critical illness. A common and clinically important problem. Crit Care Clin 1991; 7:225–252.
13. Flakeb G, Villarread D, Chapman D. Is hypokalemia a cause of ventricular arrhythmias? J Crit Illness 1986; 1:66–74.
14. Stanaszek WF, Romankiewicz JA. Current approaches to management of potassium deficiency. Drug Intell Clin Pharm 1985; 19:176–184.
15. Trissel LA. Handbook on Injectable Drugs. 13th ed. Bethesda, MD: Amer Soc Health System Pharmcists, 2005; 1230.
16. Kruse JA, Carlson RW. Rapid correction of hypokalemia using concentrated intravenous potassium chloride infusions. Arch Intern Med 1990; 150:613–617.
17. Kim GH, Han JS. Therapeutic approach to hypokalemia. Nephron 2002; 92 Suppl 1:28–32.
18. Whang R, Flink EB, Dyckner T, et al. Magnesium depletion as a cause of refractory potassium repletion. Arch Intern Med 1985; 145:1686–1689.
Hyperkalemia
19. Williams ME. Endocrine crises. Hyperkalemia. Crit Care Clin 1991; 7:155–174.
20. Evans KJ, Greenberg A. Hyperkalemia: a review. J Intensive Care Med 2005; 20:272–290.
21. Rimmer JM, Horn JF, Gennari FJ. Hyperkalemia as a complication of drug therapy. Arch Intern Med 1987; 147:867–869.
22. Don BR, Sebastian A, Cheitlin M, et al. Pseudohyperkalemia caused by fist clenching during phlebotomy. N Engl J Med 1990; 322:1290–1292.
23. Howard SC, Jones DP, Pui C-H. The tumor lysis syndrome. N Engl J Med 2012; 364:1844–1854.
24. Krisanda TJ. Digitalis toxicity. Postgrad Med 1992; 91:273–284.
25. Ponce SP, Jennings AE, Madias N, Harington JT. Drug-induced hyperkalemia. Medicine 1985; 64:357–370.
26. Perazella MA. Drug-induced hyperkalemia: old culprits and new offenders. Am J Med 2000; 109:307–314.
27. Palmer BF. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
28. Oster JR, Singer I, Fishman LM. Heparin-induced aldosterone suppression and hyperkalemia. Am J Med 1995; 98:575–586.
29. Greenberg S, Reiser IW, Chou SY, et al. Trimethoprim-sulfamethoxazole induces reversible hyperkalemia. Ann Intern Med 1993; 119:291–295.
30. Vraets A, Lin Y, Callum JL. Transfusion-associated hyperkalemia. Transfus Med Rev 2011; 25:184–196.
31. Aboudara MC, Hurst FP, Abbott KC, et al. Hyperkalemia after packed red blood cell transfusion in trauma patients. J Trauma 2008; 64:S86–S91.
32. Abu-Hamden DK, Sondheimer JH, Mahajan SK. Cautopyeiophagia: cause of life-threatening hyperkalemia in a patient undergoing hemodialysis. Am J Med 1985; 79:517–519.
33. Weisberg L. Management of severe hyperkalemia. Crit Care Med 2008; 36:3246–3251.
34. Tran HA. Extreme hyperkalemia. South Med J 2005; 98:729–732.
35. Bosnjak ZJ, Lynch C, 3rd. Cardiac Electrophysiology. In: Yaksh T, et al., eds. Anesthesia: Biologic Foundations. New York: Lippincott-Raven, 1998; 1001–1040.
36. Clausen T, Everts ME. Regulation of the Na, K-pump in skeletal muscle. Kidney Int 1989; 35:1–13.
37. Harel Z, Harel S, Shah PS, et al. Gastrointestinal adverse events with sodium polystyrene sulfonate (Kayexalate) use: a systematic review. Am J Med 2013; 126:264.e9–264.e24.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


